 作者投稿
作者投稿 专家审稿
专家审稿 编辑办公
编辑办公 主编办公
主编办公
Quantitative Imaging Study of Liver and Spleen Lesions in Patients with Gaucher Disease
-
摘要:目的
通过对治疗后戈谢病患者的肝脾系统病变进行定量影像学评估,以加深对戈谢病的了解。
方法自1999年8月至2018年8月对来首都医科大学附属北京儿童医院就诊的戈谢病患儿进行登记和临床检查治疗,并对2018年8月来本院集中随诊的40例戈谢病患儿进行定量影像学研究,同时匹配了34名正常志愿者。所有受试者行磁共振成像扫描。并对受试者肝脾的脂肪分数(FF)、铁含量(R2*)、标准表观扩散系数(sADC)、慢速表观扩散系数(D)、快速表观扩散系数(D*)和灌注分数(f)等参数进行测量。对戈谢病患者和正常者所测得的定量参数值采用独立样本t检验。
结果戈谢病患者的肝脾的脂肪分数(FF)、铁含量(R2*)、标准表观扩散系数(sADC)、慢速表观扩散系数(D)、快速表观扩散系数(D*)和灌注分数(f)等参数无显著性差异,肝脏弹性值也在正常范围内,但患者的肝脾体积与正常者呈明显差异。
结论治疗后戈谢病患者除肝脾体积仍大于正常人,其余定量参数在正常范围内,说明长期的酶替代治疗可在一定程度上延缓肝脾病变的进展,定量影像学在戈谢病评估中具有一定的价值。
Abstract:ObjectiveQuantitative imaging evaluation was performed on the liver and spleen system lesions of patients with Gaucher disease after treatment. in order to deepen the understanding of Gaucher disease.
MethodsFrom August 1999 to August 2018, we registered, examined and treated children with Gaucher disease, and conducted quantitative imaging research on 40 children with Gaucher disease who were intensively followed up in Beijing Children's hospital, Capital Medical University until August 2018. At the same time, 34 normal volunteers were matched. All subjects were scanned with magnetic resonance imaging(MRI). The fat fraction(FF), iron content(R2*), standard apparent diffusion coefficient(sADC), slow apparent diffusion coefficient(D), fast apparent diffusion coefficient(D*) and perfusion fraction(f)of the liver and spleen were measured. The quantitative parameter values measured by patients with Gaucher disease and normal subjects were statistically analyzed by independent sample t-tests.
ResultsThe results showed that there was no significant difference in FF, R2*, sADC, D, D*, f of the liver and spleen, and liver elasticity was also within the normal range. However, the volume of liver and spleen in patients was significantly different from that in normal subjects.
ConclusionsAfter treatment, the volume of the liver and spleen in patients with Gaucher disease is greater than that of normal people, but other quantitative parameters are within the normal range, indicating that long-term enzyme replacement therapy can delay the progress of liver and spleen diseases to a certain extent. Quantitative imaging has a certain value in the evaluation of Gaucher disease.
-
Keywords:
- gaucher disease /
- pediatric /
- MRI
-
戈谢病是一种罕见的常染色体隐性遗传性脂质代谢障碍疾病。一般认为溶酶体中编码葡萄糖脑苷脂酶的结构基因变异致葡萄糖脑苷脂酶缺乏,其底物葡萄糖脑苷脂不能及时被分解,从而在巨噬细胞溶酶体中积聚,最终形成典型的戈谢细胞[1]。肝脾是最易受累的器官,呈进行性和持续性增大改变[2]。若损伤因子不去除,会增加肝纤维化、肝硬化和门脉高压的风险[3]。脾功能亢进可导致全血细胞减少、微循环异常等,临床表现为贫血和铁蛋白血症[4]。本文旨在利用定量影像学的方法探索戈谢肝脾病变,为后续的治疗提供一定的价值。
1. 资料与方法
1.1 一般资料
首都医科大学附属北京儿童医院为国内首家集中诊治戈谢病患儿的儿童医院,自1999年8月至2018年8月对来本院就诊的戈谢病患儿进行登记和临床检查治疗,并对2018年8月来院集中随诊的戈谢病患儿进行定量影像学研究。选取Ⅰ型戈谢病患者40例为病例组,其中男23例,女17例,年龄10~34岁,平均(20.75±5.46)岁(随访年龄)。所有患者均符合戈谢病诊断标准[5],即骨髓检查发现戈谢细胞、白细胞或皮肤成纤维细胞中葡萄糖脑苷酶的活性比正常人低30%,临床主要表现为肝脾肿大、贫血及血小板减少。所有患者确诊后便开始长期酶替代疗法治疗,有15例患者行脾切除术。在本研究中,所有患者均肝功能等生化检查,结果基本在正常范围内。此外,另招募了34名正常志愿者为正常组,其中男20名,女14名,年龄15~32岁,平均(22.41±4.73)岁。病例组排除其他系统性疾病,如尼曼匹克病、海蓝组织细胞增生症等。正常组纳入标准为既往体检正常或全身MRI检查无异常。该研究经首都医科大学附属北京儿童医院医学伦理委员会批准(审批号:[2022]-E-022-R),所有受试者均签署知情同意书。
1.2 方法
在3.0T MRI机器(GE Discovery MR 750)上进行检查。在开始扫描之前,所有受试者都进行呼气末屏气约20 s训练。磁共振各扫描序列见表 1。定量非对称回波的最小二乘估算法迭代水脂分离(the iterative decomposition of water and fat with echo asymmetry and least-squares estimation quantitation,IDEAL-IQ)序列扫描范围包括肝、脾全轴面,自动计算翻转角度,以消除T1对脂肪分量计算的影响;此外,体素内不相干运动(the intravoxel incoherent motion imag-ing,IVIM)序列,共有10个不同的b值被设定分别为0、50、100、150、200、400、600、800、1000、1200 s/mm2,以确保结果的准确性并增加表面扩散系数(apparent diffusion coefficient,ADC)重复性;肝脏容积加速采集(liver acquisition with volume acceleration,LAVA)序列扫描范围覆盖整个肝脏,屏气时间约21 s;磁共振弹力成像(magnetic resonance elastography,MRE)常规序列包括横断面T1WI,横断面T2WI加脂肪抑制序列,冠状面稳态进动快速成像(fast imaging employing steady state acquisition,FIESTA)序列,振动频率为60 Hz,屏气扫描约28 s,获得1层,触发4次,选择肝门和肝上两层作为扫描层,上下两层用适当的区域饱和。
表 1 磁共振各扫描序列参数Table 1. MRI scanning sequence parameters序列/参数 TR(ms) TE(ms) FOV(mm2) 矩阵 层厚(mm) 层间距 IDEAL 7.2 min full 360×288 160×160 8 1.0 IVIM 2000 53.5 240×240 256×256 5 1.5 LAVA 3.3~3.8 1.5~1.8 360×360~400×400 288×200 5 2.5 MRE 50 24 360×360 N 8 1.0 结束扫描后,采集的数据被传输到AW4.6工作站由一名接受过后期处理培训的儿科放射科医生进行后处理。脂肪分数(fat fraction,FF)图和R2*图是根据IDEAL-IQ序列自动生成的,R2*图可测定铁含量值,单位赫兹(Hz)。使用functool软件包处理IVIM,计算扩散和灌注相关参数。b为扩散敏感因子,将b=200 s/mm2作为临界值,高于临界值表示弥散参数,低于临界值表示灌注参数。人工绘制ROI,包括所有受试者的FF图、R2*图、sADC、D、D*和f参数图上的整个肝脏和脾脏。记录相应的参数,并将最终结果作为三次测量的平均值。sADC为所有b值的平均ADC;D为纯水分子扩散,单位mm2/s;D*为体素中微循环的非相干运动,单位mm2/s;f表示微循环灌注效应扩散在总扩散效应中的体积比。LAVA成像可用于进行多平面重建,手动逐层绘制肝脾轮廓,然后计算肝脾体积。MRE扫描获得相应振幅和波形图,弹性图由专业处理软件通过拟合算法生成。肝脏的感兴趣区(region of interest,ROI)符合:(1) 选择固定体积ROI以确保ROI的准确性;(2)弹性区内ROI颜色均匀;(3)波形上的ROI应保证振幅良好,传播方向清晰,无损伤;(4) 振幅图的主要解剖结构是肝实质,尽量避免肝缘、大血管、胆道、伪影和脂肪变性。
1.3 统计学分析
使用SPSS 17.0统计学软件进行分析。通过独立样本t检验评估病例组和正常组之间的参数差异;测量数据以(x±s)表示,P<0.05为统计学显著性阈值。
2. 结果
所有受试者均获得良好的FF和R2*图像,即高分辨率和无伪影。病例组和正常组肝脾的FF和R2*结果见表 2,两组之间的统计数据比较无显著差异。IVIM图像以红色、黄色、绿色和蓝色表示,表示水分子扩散和微循环灌注,红色表示高灌注和低水分子扩散限制,蓝色表示低灌注和高水分子扩散限制,黄色和绿色表示中间状态。病例组和正常组肝脾sADC、D、D*和f值见表 3,两组间所有参数比较,差异均无统计学意义(P>0.05)。图 1显示了典型戈谢病患者和正常志愿者的肝脾体积图像。去除15例行脾切除术的患者,病例组和正常组的肝脾脏体积均存在显著差异,见表 4。同时,所有受试者均获得了高分辨率且无伪影的MRE。一般来说,随着肝脏硬度的增加,波形的波长逐渐增加,弹性图的颜色由蓝色变为绿色、黄色和红色。正常肝脏可见连续波传播波形。戈谢病患者的平均肝脏弹性值为(2.58±0.83)kPa,正常肝脏的弹性值为(2.35±0.34)kPa[6]; 两组之间无显著差异(P=0.100)。
表 2 两组间FF和R2*比较(x±s)Table 2. Comparison of FF and R2* between the two groups(x±s)组别 n FF(%) R2*(Hz) 肝脏 脾脏 肝脏 脾脏 病例组 40 1.84±0.50 2.01±0.77 54.18±24.13 39.17±26.32 正常组 34 2.02±0.46 1.79±0.34 49.45±7.96 42.32±19.84 t值 1.273 1.119 1.712 0.311 P值 0.643 0.270 0.093 0.757 表 3 两组间各IVIM参数比较(x±s)Table 3. Comparison of IVIM parameters between the two groups(x±s)组别 n 肝脏 脾脏 sADC×10-3
(mm2/s)D×10-3
(mm2/s)D*×10-1
(mm2/s)f值 sADC×10-3
(mm2/s)D×10-3
(mm2/s)D*×10-1
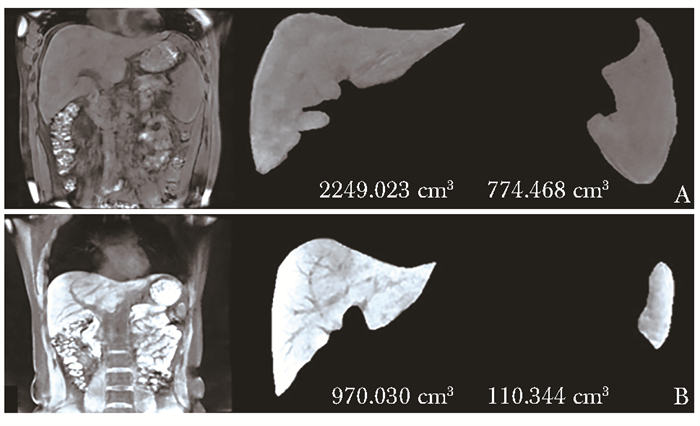
(mm2/s)f值 病例组 40 1.42±0.35 1.02±0.53 0.94±0.45 0.32±0.11 0.83±0.13 0.66±0.15 0.57±0.42 0.20±0.09 正常组 34 1.50±0.21 0.99±0.29 1.02±0.49 0.31±0.08 0.84±0.09 0.67±0.13 0.66±0.44 0.17±0.07 t值 -1.199 0.257 -0.730 0.320 -0.441 -0.463 -0.750 1.400 P值 0.234 0.798 0.468 0.750 0.662 0.645 0.456 0.167 ![]() 图 1 患者肝脾影像学检查结果A.一男性患者肝脾体积增大,肝脾体积分别为2249.023 cm3、774.468 cm3;B.一健康女性肝脾体积分别为970.030 cm3、110.344 cm3Figure 1. Results of liver and spleen imaging examination表 4 两组间肝脾体积比较(x±s, cm3)Table 4. Comparison of the liver and spleen volume between the two groups(x±s, cm3)
图 1 患者肝脾影像学检查结果A.一男性患者肝脾体积增大,肝脾体积分别为2249.023 cm3、774.468 cm3;B.一健康女性肝脾体积分别为970.030 cm3、110.344 cm3Figure 1. Results of liver and spleen imaging examination表 4 两组间肝脾体积比较(x±s, cm3)Table 4. Comparison of the liver and spleen volume between the two groups(x±s, cm3)组别 n 肝脏 脾脏 病例组 40 1498.34±452.94 411.46±228.01 正常组 34 1308.20±320.23 218.74±88.57 t值 2.050 4.150 P值 0.044 0.000 3. 讨论
戈谢病是一种罕见疾病,在全球范围内发病率较低,为1/60 000~1/40 000[7-8]。由于该病发病率低,因此对其了解有限。目前,主要认为葡萄糖脑苷脂在单核-巨噬细胞系统中的积累、各种因素对巨噬细胞的激活等都参与了戈谢病的生理过程[9]。
肝脾肿大是一种常见的表现,尤其是Ⅰ型戈谢病患者。它通常在儿童时期发展。脾脏肿大比肝脏肿大更明显,占戈谢病患者的95%[10]。扩大的肝脏和脾脏会对邻近器官和血管产生影响。病理检查显示肝中央静脉、肝静脉和脾静脉周围有大量的戈谢细胞。测量肝脏和脾脏体积有许多成像方法,LAVA是一种快速三维容积T1加权脂肪抑制成像技术。本研究发现,治疗后戈谢病患者的肝脾体积仍大于正常人,脾肿大更为明显。肝脏体积可以直接反映肝实质细胞的总量,是肝脏储备功能的重要指标,与肝脏的许多病理变化密切相关。值得注意的是,肝脾肿大是戈谢病最突出的表现,并且是持续性和进行性的。这些患者仍需继续接受长期酶替代疗法治疗。在酶替代疗法治疗前,15例戈谢病患者因严重脾肿大和明显的脾功能亢进而接受脾切除术以缓解症状。然而,脾切除术会进一步增加肝脏体积并引起其他并发症,这也是需要考虑的因素。当然,不排除在测量肝脏和脾脏体积时,人为误差也可能对结果产生一定影响。
随着葡萄糖脑苷脂在肝脏中的积累,它将增加肝纤维化和肝硬化的风险[11]。因此,肝纤维化和肝硬化作为戈谢病的严重并发症之一,需要引起更多的关注。此时,有必要密切监测肝脏的弹性值,以反映肝脏的发育和治疗效果的评估。MRE根据扫描部位组织的硬度和弹性,通过机械波实现“图像触诊”,也可用于心脏、大脑、乳房等部位[12-15]。弹力图颜色由“蓝-绿-橙-红”代表“器官硬度加重”。正常人的肝脏呈蓝色或微绿色,而肝纤维化患者的肝脏呈黄色或橙色,肝硬化患者的肝脏通常呈红色。正常人的波形图显示波形传播良好,波数正常。在肝纤维化或肝硬化患者中,波形传输可能中断,波形数量减少。在本研究中,戈谢病患者肝脏的平均弹性值为(2.58±0.83)kPa,正常人的平均弹性值为(2.35±0.34)kPa[6],两组在统计学上比较无显著差异。国外报道的肝脏弹性值为2.3~2.8 kPa[16],与国内报道相重叠。本研究结果显示,戈谢病患者肝纤维化程度较轻,不符合肝纤维化影像学诊断标准。此外,经过多年的酶替代疗法治疗,它在一定程度上延缓了肝病的进展。在其他方面,MRE的后处理过程复杂,人为因素引起的测量差异也可能影响最终结果。
葡萄糖脑苷脂可能是肝脾脂质成分动态失衡的主要原因。同时,当葡萄糖脑苷脂沉积在肝脾血管中时,血管会被堵塞,最终导致肝脾梗死,高铁血红蛋白含量增加。因此,有必要监测戈谢病患者肝、脾的脂肪含量和铁含量。IDEAL-IQ作为一种新的定量技术,将定性水脂肪分离改进为定量脂肪比测量。一次屏气扫描可获得六种组织器官图像,即水相、脂肪相、同相、反相、脂肪信号成分图和R2*图[17]。本研究中40例患者均接受了酶替代疗法治疗,同时设立了34名正常对照组。由于这些患者在诊断后、治疗前没有进行定量影像学评估。因此,本文不能垂直评估与治疗相关的时间变化,只能进行横断面评估。本研究结果表明,患者和正常人之间的肝脏和脾脏FF和铁含量无显著的统计学差异。同时结合临床肝功能检测,患者肝脏生化指标基本在正常范围内。一般来说,戈谢病患者肝脾巨噬细胞中葡萄糖脑苷脂的积累会引起肝脾的病理生理和临床改变。为此,本组患者接受酶替代疗法治疗已有多年。酶替代疗法通过阻断葡萄糖脑苷酶的沉积来减少器官和组织中葡萄糖脑苷的负荷,并且未进展到肝纤维化甚至肝硬化的程度。在一定程度上说明了酶替代疗法的有效性,对疾病的治疗有一定的延缓和改善作用。当然,酶替代疗法的效果也受到治疗开始时间、不可逆并发症的存在和酶剂量的影响。另一方面,骨骼系统受累是戈谢病的严重并发症[18]。与骨骼病变相比,此时肝脾疾病的程度远低于骨骼系统。个体差异是另一个因素,如性别和年龄[19]。本研究结果还显示,病例组和正常组之间的R2*无显著差异。在本研究收集的患者中,除肝脾肿大外,常规腹部MRI没有其他异常表现,定量成像提供的定量信息也在正常范围内。戈谢病患者可存在红细胞生成障碍和铁代谢缺陷引起的贫血和高铁蛋白血症。酶替代疗法可以释放铁,改善铁状态,纠正贫血。在一些研究中,已观察到酶替代疗法治疗数月后血清铁蛋白浓度下降,并被认为是疾病活动的标志[4]。因此,铁含量的测量可为评估疾病严重程度和治疗反应提供额外的定量信息。
戈谢细胞的积累也会引起水分子扩散和组织器官微循环灌注的变化。但过去弥散加权成像(diffusion weighted imaging,DWI)受血液灌注的影响,不能真实反映组织中水分子的扩散[20-22]。基于DWI扩展的IVIM技术可以通过双指数模型计算将水分子扩散与微循环灌注分离,更准确地量化组织扩散和微循环灌注信息[23-25]。结果表明,病例组和正常组之间的sADC、D、D*和f值无显著差异。然而,在数据中发现了一些细微的差异。病例组肝脏的D和f值略高于正常组,而D*值略低于正常组。病例组脾脏D值和D*值略低于正常组,f值略高于正常组。这些差异也是可以预料的,脾脏肿大比肝脏更明显,戈谢细胞的增加阻碍了水分子的扩散。与肝脏相比,脾脏的D值略低于正常组。同时,脾脏梗死的发生率远高于肝脏。由于梗死的发生,脾脏灌注分数可能降低。由于代偿效应,与灌注相关的弥散运动将增加。值得注意的是,f值不仅受血管微循环的影响,还受其他因素(如液体流量)的影响。f可能偏离实际水平。有研究发现慢性肝病患者和正常志愿者之间的ADC值无差异[26]。而据报道,D值和ADC值在肝脏良恶性局灶性病变的研究中有显著差异。Chen等[27]提出,肝纤维化患者的D*和f值明显低于正常肝脏。研究结果多样化的原因可能如下:(1)虽然肝脏和脾脏系统是最常见的受累器官,但本研究收集的戈谢病患者的肝脏和脾脏受累程度较少。这些患者自从被诊断出患有该病以来,已经接受了很长时间的酶替代疗法治疗,这在一定程度上延缓了疾病的进展;(2) 不同研究机构设置的IVIM扫描参数不同,如b的数量和分布不同。戈谢病的发病机制复杂多样。对各种现象和结果的具体原因没有全面的了解,需要进一步探讨。
综上,由于戈谢病患者体内葡萄糖脑苷脂的积累,最有可能累及肝脏和脾脏,增加肝纤维化和肝硬化的风险。在本研究中,定量MRI用于评估戈谢病患者的肝脾病变。发现戈谢病患者的肝脾体积与正常者有显著差异,尤其是脾肿大。然而,治疗后的戈谢病患者和正常志愿者在FF、铁含量、水分子扩散、微循环灌注和肝弹性方面无显著性差异,这表明肝脾肿大是戈谢病最突出的表现,并且是持续性和进行性的。酶替代疗法可在一定程度上延缓肝脾病变的进展。定量MRI的应用对临床的后续治疗以及疾病的进展的评估有一定的价值。本研究存在一些局限性,如缺乏治疗前基线数据。因此,需要进一步增加对临床资料的重视,并在后续研究中更深入探索。
作者贡献:李笛、彭芸、陶晓娟、张宁宁、霍爱华、胡迪、吕艳秋、张永红参与选题和设计,参与资料的分析与解释以及数据的获得;李笛、彭芸、陶晓娟修改论文中关键性理论或其他主要内容;彭芸对最终要发表的论文版本进行了审阅和把关。利益冲突:所有作者均声明不存在利益冲突。 -
![]()
图 1 患者肝脾影像学检查结果
A.一男性患者肝脾体积增大,肝脾体积分别为2249.023 cm3、774.468 cm3;B.一健康女性肝脾体积分别为970.030 cm3、110.344 cm3
Figure 1. Results of liver and spleen imaging examination
表 1 磁共振各扫描序列参数
Table 1 MRI scanning sequence parameters
序列/参数 TR(ms) TE(ms) FOV(mm2) 矩阵 层厚(mm) 层间距 IDEAL 7.2 min full 360×288 160×160 8 1.0 IVIM 2000 53.5 240×240 256×256 5 1.5 LAVA 3.3~3.8 1.5~1.8 360×360~400×400 288×200 5 2.5 MRE 50 24 360×360 N 8 1.0  下载: 导出CSV
下载: 导出CSV
表 2 两组间FF和R2*比较(x±s)
Table 2 Comparison of FF and R2* between the two groups(x±s)
组别 n FF(%) R2*(Hz) 肝脏 脾脏 肝脏 脾脏 病例组 40 1.84±0.50 2.01±0.77 54.18±24.13 39.17±26.32 正常组 34 2.02±0.46 1.79±0.34 49.45±7.96 42.32±19.84 t值 1.273 1.119 1.712 0.311 P值 0.643 0.270 0.093 0.757
下载: 导出CSV
表 3 两组间各IVIM参数比较(x±s)
Table 3 Comparison of IVIM parameters between the two groups(x±s)
组别 n 肝脏 脾脏 sADC×10-3
(mm2/s)D×10-3
(mm2/s)D*×10-1
(mm2/s)f值 sADC×10-3
(mm2/s)D×10-3
(mm2/s)D*×10-1
(mm2/s)f值 病例组 40 1.42±0.35 1.02±0.53 0.94±0.45 0.32±0.11 0.83±0.13 0.66±0.15 0.57±0.42 0.20±0.09 正常组 34 1.50±0.21 0.99±0.29 1.02±0.49 0.31±0.08 0.84±0.09 0.67±0.13 0.66±0.44 0.17±0.07 t值 -1.199 0.257 -0.730 0.320 -0.441 -0.463 -0.750 1.400 P值 0.234 0.798 0.468 0.750 0.662 0.645 0.456 0.167
下载: 导出CSV
表 4 两组间肝脾体积比较(x±s, cm3)
Table 4 Comparison of the liver and spleen volume between the two groups(x±s, cm3)
组别 n 肝脏 脾脏 病例组 40 1498.34±452.94 411.46±228.01 正常组 34 1308.20±320.23 218.74±88.57 t值 2.050 4.150 P值 0.044 0.000
下载: 导出CSV
-
[1] Zion YC, Papadopulos E, Wajnrajch M, et al. Rethinking fatigue in Gaucher disease[J]. Orphanet J Rare Dis, 2016, 11: 53. doi: 10.1186/s13023-016-0435-x
[2] Stirnemann J, Belmatoug N, Camou F, et al. A review of gaucher disease pathophysiology, clinical presentation and treatments[J]. Int J Mol Sci, 2017, 18: 441. doi: 10.3390/ijms18020441
[3] Adar T, Ilan Y, Elstein D, et al. Liver involvement in Gaucher disease-Review and clinical approach[J]. Blood Cells Mol Dis, 2018, 68: 66-73. doi: 10.1016/j.bcmd.2016.10.001
[4] Nguyen Y, Stirnemann J, Belmatoug N. Gaucher disease: A review[J]. Rev Med Interne, 2019, 40: 313-322. doi: 10.1016/j.revmed.2018.11.012
[5] Koppe T, Doneda D, Siebert M, et al. The prognostic value of the serum ferritin in a southern Brazilian cohort of patients with Gaucher disease[J]. Genet Mol Biol, 2016, 39: 30-34. doi: 10.1590/1678-4685-GMB-2015-0125
[6] 石喻, 郭启勇, 张兰, 等. 3.0TMR弹性成像评价健康者及慢性肝病患者肝弹性值的初步研究[J]. 中华放射学杂志, 2013, 47: 1005-1008. doi: 10.3760/cma.j.issn.1005-1201.2013.11.012 [7] Feng Y, Huang Y, Tang C, et al. Clinical and molecular characteristics of patients with Gaucher disease in southern China[J]. Blood Cells Mol Dis, 2018, 68: 30-34. doi: 10.1016/j.bcmd.2016.10.026
[8] Kawasaki H, Suzuki T, Ito K, et al. Minos-insertion mutant of the Drosophila GBA gene homolog showed abnormal phenotypes of climbing ability, sleep and life span with accumulation of hydroxyl-glucocerebroside[J]. Gene, 2017, 614: 49-55. doi: 10.1016/j.gene.2017.03.004
[9] Gary SE, Ryan E, Steward AM, et al. Recent advances in the diagnosis and management of Gaucher disease[J]. Expert Rev Endocrinol Metab, 2018, 13: 107-118. doi: 10.1080/17446651.2018.1445524
[10] Mehta A, Belmatoug N, Bembi B, et al. Exploring the patient journey to diagnosis of Gaucher disease from the perspective of 212 patients with Gaucher disease and 16 Gaucher expert physicians[J]. Mol Genet Metab, 2017, 122: 122-129. doi: 10.1016/j.ymgme.2017.08.002
[11] 石俊英, 张斯佳, 史景璐, 等. 乙型肝炎相关性肝纤维化比较分析[J]. 中国临床医学影像杂志, 2017, 28: 447-449. doi: 10.3969/j.issn.1008-1062.2017.06.018 [12] Manduca A, Bayly PJ, Ehman RL, et al. MR elastography: principles, guidelines, and terminology[J]. Magn Reson Med, 2021, 85: 2377-2390. doi: 10.1002/mrm.28627
[13] Chartrain AG, Kurt M, Yao A, et al. Utility of preopera-tive meningioma consistency measurement with magnetic resonance elastography (MRE): a review[J]. Neurosurg Rev, 2019, 42: 1-7. doi: 10.1007/s10143-017-0862-8
[14] Varyani F, Samuel S. Can magnetic resonance enterography (MRE) replace ileo-colonoscopy for evaluating disease activity in Crohn's disease?[J]. Best Pract Res Clin Gastroenterol, 2019, 38-39: 101621. doi: 10.1016/j.bpg.2019.05.008
[15] Grand DJ, Deepak P, Rimola J. MRE evaluation of intestinal inflammation: qualitative and quantitative assessment[J]. Top Magn Reson Imaging, 2021, 30: 13-22. doi: 10.1097/RMR.0000000000000270
[16] Rustogi R, Horowitz J, Harmath C, et al. Accuracy of MR elastography and anatomic MR imaging features in the diagnosis of severe hepatic fibrosis and cirrhosis[J]. J Magn Reson Imaging, 2012, 35: 1356-1364. doi: 10.1002/jmri.23585
[17] Eskreis-Winkler S, Corrias G, Monti S, et al. IDEAL-IQ in an oncologic population: meeting the challenge of concomitant liver fat and liver iron[J]. Cancer Imaging, 2018, 18: 51. doi: 10.1186/s40644-018-0167-3
[18] Hughes D, Mikosch P, Belmatoug N, et al. Gaucher disease in bone: from pathophysiology to practice[J]. J Bone Miner Res, 2019, 34: 996-1013. doi: 10.1002/jbmr.3734
[19] Baum T, Yap SP, Dieckmeyer M, et al. Assessment of whole spine vertebral bone marrow fat using chemical shift-encoding based water fat MRI[J]. J Magn Reson Imaging, 2015, 42: 1018-1023. doi: 10.1002/jmri.24854
[20] Kele PG, van der Jagt EJ. Diffusion weighted imaging in the liver[J]. World J Gastroenterol, 2010, 16: 1567-1576. doi: 10.3748/wjg.v16.i13.1567
[21] Roder C, Haas P, Tatagiba M, et al. Technical limitations and pitfalls of diffusion-weighted imaging in intraoperative high-field MRI[J]. Neurosurg Rev, 2021, 44: 327-334. doi: 10.1007/s10143-019-01206-0
[22] Wichtmann BD, Zöllner FG, Attenberger UI, et al. Multiparametric MRI in the diagnosis of prostate cancer: Physical foundations, limitations, and prospective advances of diffusion-weighted MRI[J]. RoFo, 2021, 193: 399-409. doi: 10.1055/a-1276-1773
[23] Le Bihan D. What can we see with IVIM MRI?[J]. NeuroImage, 2019, 187: 56-67. doi: 10.1016/j.neuroimage.2017.12.062
[24] Fadnavis S, Endres S, Wen Q, et al. Bifurcated topological optimization for IVIM[J]. Front Neurosci, 2021, 15: 779025. doi: 10.3389/fnins.2021.779025
[25] Vasylechko SD, Warfield SK, Afacan O, et al. Self-supervised IVIM DWI parameter estimation with a physics based forward model[J]. Magn Reson Med, 2022, 87: 904-914. doi: 10.1002/mrm.28989
[26] Watanabe H, Kanematsu M, Goshina S, et al. Characterizing focal hepatic lesions by free breathing intravoxel incoherent motion MRI at 3.0 T[J]. Acta Radiol, 2014, 55: 1166-1173. doi: 10.1177/0284185113514966
[27] Chen C, Wang B, Shi D, et al. Initial study of biexponential model of intravoxel incoherent motion magnetic resonance imaging in evaluation of the liver fibrosis[J]. Chin Med J, 2014, 127: 3082-3087.
计量
- 文章访问数: 693
- HTML全文浏览量: 90
- PDF下载量: 50


