 作者投稿
作者投稿 专家审稿
专家审稿 编辑办公
编辑办公 主编办公
主编办公
A Case Report of Von Hippel-Lindau Syndrome with Hypertension and Diabetes as the First Manifestation
-
摘要:
冯·希佩尔-林道(VHL)综合征,又称脑视网膜血管瘤病,属于神经内分泌肿瘤的一种。发病率较高,且遗传性极高,可涉及视网膜及中枢神经系统、各个脏器和各种组织部位等。本文报道1例以高血压、糖尿病为主要表现的VHL综合征,旨在提高对本病的认识,减少误诊和漏诊。
-
关键词:
- 冯·希佩尔-林道综合征 /
- 高血压 /
- 糖尿病
Abstract:Von Hippel-Lindau (VHL) syndrome, also known as cerebral retinal angiomatosis, is a kind of neuroendocrine tumor. The incidence rate is high, and the heredity is very high, which can involve the retina, central nervous system, various organs and various tissue parts. This paper reports a case of VHL syndrome with hypertension and diabetes as the main manifestations, in order to improve the understanding of the disease and reduce misdiagnosis and missed diagnosis.
-
Keywords:
- von Hippel-Lindau syndrome /
- hypertension /
- diabetes
-
冯·希佩尔-林道综合征(von Hippel-Lindau syndrome),又称VHL综合征,或脑视网膜血管瘤病,是一种罕见的因位于染色体3P25.3的VHL抑癌基因发生突变所致的常染色体显性遗传性疾病,表现为血管母细胞瘤累及小脑、脊髓、肾脏及视网膜,病变包括肾脏血管瘤、肾细胞癌及嗜铬细胞瘤等[1]。该病发病率为1/85 000~1/36 000,65岁时外显率>90%,子女发病率为50%,主要表现为同一家族内不同成员患有部位及组织学各不相同的各种肿瘤,可涉及视网膜及中枢神经系统、内脏和生殖系统等[2]。本文报道1例2016年就诊于重庆医科大学附属第一医院,以高血压、糖尿病为主要表现,肿瘤病变累及肾脏、肾上腺及胰腺等部位的患者。从该患者的临床资料及相关文献复习,以期帮助临床医生更好地认识该疾病。
1. 临床资料
患者女性,32岁。因“发现血压升高1年,加重伴多饮、多食、消瘦1月”于重庆医科大学附属第一医院(以下简称我院)就诊。患者2015年7月于妊娠39周发现血压升高,收缩压最高180 mm Hg。自诉产后约3周后血压自行恢复正常,未服用药物。2016年5月再次发现血压升高伴无明显诱因出现多饮、多食、多尿,并体重减轻(下降约3 kg),测血糖、血压升高,于外院完善相关影像学检查(见辅助检查)后考虑特殊类型糖尿病、继发性高血压,予以二甲双胍片降血糖、硝苯地平控释片降血压等对症治疗后症状好转,现为进一步明确病因就诊于我院。患者发病以来饮食、小便如上述,睡眠及大便正常。既往有慢性乙型病毒性肝炎病史10年,未诊治。父亲有高血压病史,因肾肿瘤去世,否认糖尿病家族史。患者存在明确家族聚集倾向,呈显性遗传:祖母早逝,死因不详;父亲有双侧肾肿瘤病史;弟弟姑姑有颅内良性肿瘤病史;姑姑的儿子(即表弟)有双侧肾上腺嗜铬细胞瘤病史,姑姑的女儿(即表妹)有双侧肾上腺嗜铬细胞瘤病史(图 1)。
体格检查:体温36.8 ℃,脉搏84次/分,呼吸16次/分,血压115/88 mm Hg,身高157 cm,体重47 kg,体重指数19.06 kg/m2。正常体型,全身皮肤无紫纹,颈、胸、心、腹查体阴性。双下肢无水肿。神经系统查体无异常。
辅助检查:(1)实验室检查。血常规示血红蛋白151 g/L。口服葡萄糖耐量试验(oral glucose tolerance test,OGTT)示空腹血糖(fasting plasma glucose,FPG) 7.9 mmol/L,餐后2 h血糖(2 hPG) 16.7 mmol/L。糖化血红蛋白(glycosylated hemoglobin, HbA1c)15%。尿常规示尿酮体(-)、尿糖(3+)。血脂示低密度脂蛋白胆固醇5.28 mmol/L。粪便常规及隐血、肝肾功能及电解质、凝血功能、输血前检查、胰岛素及C肽释放试验、垂体相关激素(生长激素、催乳素、雌激素、孕激素相关激素)、甲状腺及甲状旁腺相关激素(骨碱性磷酸酶、降钙素)、血清钙、血清磷、血清钾、促肾上腺皮质激素及皮质醇昼夜节律、立卧位肾素及醛固酮无异常。
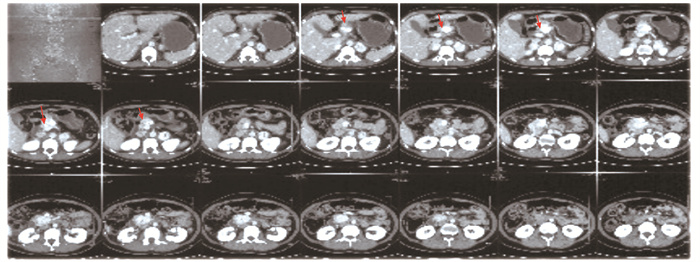
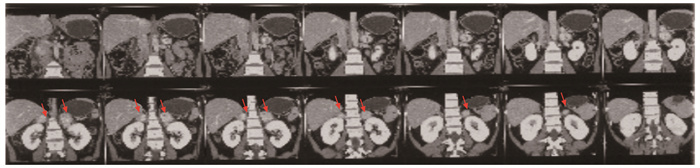
(2) 影像学检查。心电图、超声心动图及胸部X线片无异常。腹部B超显示轻度脂肪肝、右侧肾上腺占位、双侧肾囊肿;甲状腺、乳腺、妇科超声及眼底检查无异常。外院影像学资料:胰腺CT增强扫描(图 2) 显示胰头及胰颈部明显强化肿块影(胰头4.2 cm×3.7 cm、胰颈2.7 cm×3.2 cm,呈分叶状),边界较清晰、血管丰富、钙化较多,提示可能为功能性占位、胰岛细胞瘤。双侧肾上腺区CT增强扫描(图 3)显示双侧肾上腺肿块影(3.0~3.5 cm),明显强化,考虑肿瘤性病变(转移瘤、嗜铬细胞瘤);右肾中份外侧小结节影(1.2 cm)密度不均匀,考虑肿瘤性病变(肾癌、双侧肾囊肿)。我院腹部CT增强扫描显示双侧肾上腺占位性病变,胰头颈部软组织肿块并多发钙化,考虑胰岛细胞瘤伴双侧肾上腺转移、双肾多发小囊肿。脑部鞍区增强MRI显示垂体中份低信号小结节影,垂体未见增大,考虑微腺瘤可能,囊肿可能性小;眼眶、小脑及脑干未见异常。颈椎、胸椎及腰椎MRI无异常。
(3) 基因检测。见表 1。进一步证实患者VHL基因有1个杂合突变:c.499C>T(编码区第499号核苷酸由胞嘧啶变异为胸腺嘧啶),导致氨基酸改变p.R167W(第167号氨基酸由精氨酸变异为色氨酸),为错义突变,该变异不属于多态性位点,在人群中发生频率极低;人类基因突变数据库(Human Gene Mutation Database,HGMD)专业版数据库已报道与VHL基因相关,该变异为致病性变异。同时分析到SDHB基因有1个杂合突变:c.725G>A(编码区第725号核苷酸由鸟嘌呤变异为腺嘌呤),导致氨基酸改变p.R242H(第242号氨基酸由精氨酸变异为组氨酸),为错义突变,该变异不属于多态性位点,在人群中发生频率极低;HGMD专业版数据库已报道与嗜铬细胞瘤相关,该变异为致病性变异。
表 1 患者基因检测结果Table 1. Genetic test results of the patient基因 染色体位置 转录本编号 外显子 核苷酸变化 氨基酸变化 致病性分析 纯合/杂合 遗传方式 疾病/表型 VHL chr3-10191506 NM_000551 exon3 c.499C>T p.R167W Pathogenic het 1.AD
2.AD
3.AR1.VHL综合征
2.嗜铬细胞瘤
3.家族性红细胞增多症2型SDHB chr1-17349143 NM_003000 exon7 c.725G>A p.R242H Pathogenic het 1.AD
2.AD
3.-
4.AD
5.AD1.多发性错构瘤综合征
2.胃肠道间质瘤
3.副神经节瘤合并胃恶性间质瘤
4.副神经节瘤4型
5.嗜铬细胞瘤AD:常染色体显性遗传;AR:常染色体隐性遗传; VHL综合征: 冯·希佩尔-林道综合征; het:杂合突变 诊断:VHL综合征,右侧肾上腺嗜铬细胞瘤、右肾透明细胞癌、双肾囊肿、胰腺肿瘤待查;特殊类型糖尿病、继发性高血压、高脂血症;乙型肝炎病毒携带者。诊断依据:患者青年发病,无明显高血压、糖尿病危险因素,我院阅片影像学提示患者多发内脏肿瘤;先证者有明确的家族史;先证者有包括肾上腺、胰腺及右肾在内的多脏器占位,既往有一过性高血压,近期再次出现血压升高(嗜铬细胞瘤可能)、血糖明显升高不伴有胰岛素及C肽释放水平受损(胰高血糖素瘤可能)。结合患者的基因检测结果,故诊断为VHL综合征。该疾病最主要是与多发性内分泌肿瘤(multiple endocrine neoplasm,MEN)(如MEN-1或MEN-2)相鉴别。因先证者缺少MEN相关肿瘤组分,如甲状旁腺瘤、垂体瘤、甲状腺髓样瘤等证据。二者鉴别主要依靠基因筛查以进一步证实。
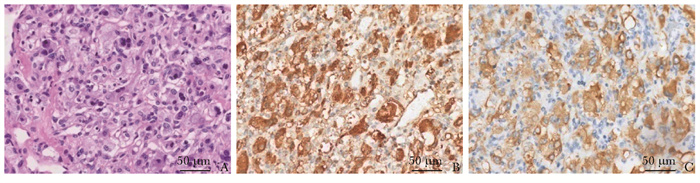
治疗:入我院后予以二甲双胍片850 mg(3次/日) 降血糖、培哚普利叔丁胺片4 mg(1次/日)+氨氯地平片5 mg (1次/日)降血压等对症治疗;监测空腹手指血糖5.4~10.4 mmol/L,餐后指血糖4.7~13.0 mmol/L,血压(93~125)/(65~94)mm Hg,心率97~110次/分。在高度怀疑该病的基础上经我院多学科讨论后制订诊疗方案:按照嗜铬细胞瘤进行扩容等术前准备,择期行右侧肾上腺及右肾肿块或结节手术治疗,后期转至胰腺专科处理胰腺肿块。按照中华医学会内分泌学分会《嗜铬细胞瘤和副神经节瘤诊断治疗的专家共识(2016版)》做术前准备,予以调整为酚苄明片10 mg(3次/日)扩容、美托洛尔片25 mg(2次/日)控制心室率,体重至术前共增加3 kg,血压为(100~ 110)/(80~90)mm Hg,心率为78~86次/分;复查血常规,血红蛋白134 g/L。患者2周后在全身麻醉下行机器人辅助右侧肾上腺肿瘤切除术+右肾部分切除术。切除组织送病理检查(图 4)显示右侧肾上腺嗜铬细胞瘤及右肾透明细胞癌,Fuhrman分级(核特征)为Ⅰ级;右肾良性囊肿。免疫组化示:①右侧肾上腺肿瘤,Ki-67个别(+),CgA(+),Syn(+),CK(-),EMA(-);②右肾肿瘤,Ki-67(+),CgA(-),Syn(-),CK (+),EMA(+),CD10(+),Vimentin(+),CK7 (+),RCC(+),CD117(-),CAIX(+),TFE-3(-)。
![]() 图 4 患者肾上腺切除组织病理结果A.(HE×400)示肿瘤细胞与正常嗜铬细胞相似,异型性不明,呈巢状排列,胞质嗜碱,呈颗粒样,核仁明显,周围可见支持细胞;B.(CgA×400)与C.(Syn×400)示肿瘤细胞CgA、Syn阳性,可鉴别肾上腺皮质肿瘤和转移性非神经内分泌肿瘤;HE:苏木精-伊红染色;CgA染色:嗜铬颗粒蛋白A免疫组化染色;Syn染色:突触素免疫组化染色Figure 4. Histopathology of adrenalectomy of the patient
图 4 患者肾上腺切除组织病理结果A.(HE×400)示肿瘤细胞与正常嗜铬细胞相似,异型性不明,呈巢状排列,胞质嗜碱,呈颗粒样,核仁明显,周围可见支持细胞;B.(CgA×400)与C.(Syn×400)示肿瘤细胞CgA、Syn阳性,可鉴别肾上腺皮质肿瘤和转移性非神经内分泌肿瘤;HE:苏木精-伊红染色;CgA染色:嗜铬颗粒蛋白A免疫组化染色;Syn染色:突触素免疫组化染色Figure 4. Histopathology of adrenalectomy of the patient治疗结果、随访及转归:术后2年随访。①左侧肾上腺占位:2017年前于当地医院行左侧肾上腺占位切除术,术后病理提示“嗜铬细胞瘤”,术后2个月复查该侧肿瘤复发,大小同术前,至今随访未增大,监测血压正常;②胰腺占位:随访肿瘤大小无变化,自行停用降血糖药1年后,近6个月来再次出现口干、多尿等症状,监测空腹手指血糖为10 mmol/L,服用二甲双胍缓释片500 mg(3次/日)降血糖,血糖控制可。
2. 讨论
VHL属于神经内分泌肿瘤的一种[3]。自1895年德国眼科医师von Hippel发现家族性视网膜血管瘤、1926年瑞典眼科医师Lindau报道视网膜血管瘤合并小脑及腹腔病变开始,直到1964年Melmom和Rosen首次命名该种家族性多发肿瘤综合征[4-6]。
研究证实,VHL综合征发生与位于3号染色体3p25-p26上的VHL抑癌基因突变相关,呈常染色体显性遗传[7]。目前认为主要分子生物学机制为:当抑癌基因VHL基因突变或缺失时,VHL蛋白异常或生成障碍,其与转录因子缺氧诱导因子1α (hypoxia-inducible factor 1 α,HIF-1α)的结合部位异常有关,导致HIF-1α增加,基因转录及生长因子上调,包括促红细胞生成素、血管内皮生长因子、血小板衍生生长因子B和参与葡萄糖摄取的其他基因等,从而激活细胞增殖和血管生成,诱发血管母细胞瘤和内脏器官肿瘤等产生[8]。同一家族内不同成员常患有部位及组织学各不相同的各种肿瘤,表现为多发性、多器官性、良恶性肿瘤共存,可涉及视网膜及中枢神经系统(血管网状细胞瘤)、内脏(胰腺肿瘤和囊肿、肾透明细胞癌和囊肿、肾上腺嗜铬细胞瘤)和生殖系统(附睾或阔韧带囊肿或囊腺瘤)等[9-10]。
统计表明,肿瘤的病理类型有60%~80%合并神经系统/视网膜血管网状细胞瘤,50%~77%合并胰腺占位(其中12%~17%为胰腺神经内分泌肿瘤),40%~50%合并肾癌(主要为透明细胞型),7%~18%合并嗜铬细胞瘤[1, 9-11]。当出现多种肿瘤特征性表现或者发现≥2个肿瘤时,需要考虑到遗传性神经内分泌肿瘤综合征。在高度怀疑该病时,应完善生化检查以评估激素水平、行影像学检查对肿瘤或增生的病灶进行定位,并进行遗传咨询和基因检测最后确诊[12-13]。
目前,VHL综合征公认的诊断标准:有中枢神经系统或视网膜血管瘤家族史,有单发血管网状细胞瘤或存在内脏实质肿瘤者;或无中枢神经系统或视网膜血管瘤家族史,有多发血管网状细胞瘤或1个血管网状细胞瘤合并内脏实质肿瘤者,需考虑该病,通过基因检测可明确诊断[3, 14-15]。该患者青年发病,无明显高血压、糖尿病危险因素,有一过性高血压及血糖明显升高[16-17]。我院阅片影像学提示患者多发内脏肿瘤;此外先证者有明确的家族史;有包括肾上腺、胰腺及右肾在内的多脏器占位。因此,患者,近期再次出现血压升高需考虑嗜铬细胞瘤。而患者血糖明显升高不伴有胰岛素及C肽释放水平受损,且有胰腺占位需考虑胰高血糖素瘤。这主要需要与多发性内分泌肿瘤病(如MEN-1/MEN-2)相鉴别。因先证者缺少MEN相关肿瘤组分,如甲状旁腺瘤、垂体瘤、甲状腺髓样瘤等证据。两者鉴别主要依靠基因筛查以进一步证实。结合患者的基因检测结果,故最终诊断为VHL综合征。
但该患者无中枢神经系统或视网膜血管瘤,考虑与VHL突变类型、突变区域和突变密码子密切相关。这些具体的突变方式可作为VHL综合征表型预测因子。突变区和突变密码子可协助定向监测VHL综合征患者病情。研究表明,VHL缺失的患者更容易发生视网膜血管瘤,而错义突变增加嗜铬细胞瘤的风险,但降低中枢神经系统血管网状细胞瘤和胰腺病变的风险;肾细胞癌在无义、移码或剪接位点突变中更常见[18-19]。外显子3突变的患者更容易发生嗜铬细胞瘤;密码子80或密码子167处的突变赋予嗜铬细胞瘤显著高于其他突变的风险[20-21]。前述先证者基因突变与这一特点比较符合,而该病患者死亡的主要原因是与肾细胞癌和中枢神经系统血管网状细胞瘤相关的并发症。在嗜铬细胞瘤尤其是复发性嗜铬细胞瘤、肾细胞癌及胃、肠、胰腺、神经内分泌肿瘤的诊疗过程中,除了散发型,均需除外遗传性疾病,如VHL综合征。
综上,本文报道了1例罕见的VHL综合征,并对该综合征的临床特点进行总结分析,为临床医生诊治该疾病提供借鉴与帮助,同时为继发性高血压及特殊类型糖尿病提供诊断思路,减少相关疾病的漏诊与误诊。
作者贡献: 蒲鹏负责收集病例资料,文章撰写并审阅修改;郑晓雅负责收集病例资料,对文章内容进行审阅修改;刘瑞闪负责收集病历资料,文章撰写;周建中负责对文章内容作批评性审阅。利益冲突:所有作者均声明不存在利益冲突。 -
![]()
图 4 患者肾上腺切除组织病理结果
A.(HE×400)示肿瘤细胞与正常嗜铬细胞相似,异型性不明,呈巢状排列,胞质嗜碱,呈颗粒样,核仁明显,周围可见支持细胞;B.(CgA×400)与C.(Syn×400)示肿瘤细胞CgA、Syn阳性,可鉴别肾上腺皮质肿瘤和转移性非神经内分泌肿瘤;HE:苏木精-伊红染色;CgA染色:嗜铬颗粒蛋白A免疫组化染色;Syn染色:突触素免疫组化染色
Figure 4. Histopathology of adrenalectomy of the patient
表 1 患者基因检测结果
Table 1 Genetic test results of the patient
基因 染色体位置 转录本编号 外显子 核苷酸变化 氨基酸变化 致病性分析 纯合/杂合 遗传方式 疾病/表型 VHL chr3-10191506 NM_000551 exon3 c.499C>T p.R167W Pathogenic het 1.AD
2.AD
3.AR1.VHL综合征
2.嗜铬细胞瘤
3.家族性红细胞增多症2型SDHB chr1-17349143 NM_003000 exon7 c.725G>A p.R242H Pathogenic het 1.AD
2.AD
3.-
4.AD
5.AD1.多发性错构瘤综合征
2.胃肠道间质瘤
3.副神经节瘤合并胃恶性间质瘤
4.副神经节瘤4型
5.嗜铬细胞瘤AD:常染色体显性遗传;AR:常染色体隐性遗传; VHL综合征: 冯·希佩尔-林道综合征; het:杂合突变  下载: 导出CSV
下载: 导出CSV
-
[1] Ben-Skowronek I, Kozaczuk S. Von Hippel-Lindau syn-drome[J]. Horm Research Paediatr, 2015, 84 (3): 145-152. doi: 10.1159/000431323
[2] Khan HA, Shahzad MA, Iqbal F, et al. Ophthalmological aspects of von-Hippel-Iindau syndrome[J]. Semin Ophthalmol, 2021, 36(7): 531-540. doi: 10.1080/08820538.2021.1897851
[3] Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease[J]. Lancet, 2003, 361(9374): 2059-2067. doi: 10.1016/S0140-6736(03)13643-4
[4] Sora S, Ueki K, Saito N, et al. Incidence of von Hippel-Lindau disease in hemangioblastoma patients: the University of Tokyo Hospital experience from 1954-1998[J]. Acta Neurochir(Wien), 2001, 143(9): 893-896. doi: 10.1007/s007010170019
[5] Huson SM, Harper PS, Hourihan MD, et al. Cerebellar haemangioblastoma and von Hippel-Lindau disease[J]. Brain, 1986, 109(Pt6): 1297-1310.
[6] de Souza Andrade J, Bambirra EA, Bicalho OJ, et al. Bila-teral papillary cystadenoma of the epididymis as a component of von Hippel-Lindau's syndrome: report of a case presenting as infertility[J]. J Urol, 1985, 133(2): 288-289. doi: 10.1016/S0022-5347(17)48920-8
[7] Rednam SP, Erez A, Druker H, et al. Von Hippel-Lindau and hereditary pheochromocytoma/paraganglioma syndromes: clinical features, genetics, and surveillance recommen-dations in childhood[J]. Clin Cancer Res, 2017, 23(12): e68-e75. doi: 10.1158/1078-0432.CCR-17-0547
[8] Hasanov E, Jonasch E. MK-6482 as a potential treatment for von Hippel-Lindau disease-associated clear cell renal cell carcinoma[J]. Expert Opinion Investig Drugs, 2021, 30(5): 495-504. doi: 10.1080/13543784.2021.1925248
[9] Friedrich CA. Von Hippel-Lindau syndrome. A pleomorphic condition[J]. Cancer, 1999, 86(11 Suppl): 2478-2482.
[10] Capogreco A, Pedicini V, Masetti C, et al. Portal hypertension determined by von Hippel Lindau syndrome[J]. Liver Int, 2021, 41(6): 1421-1422. doi: 10.1111/liv.14783
[11] Chou A, Toon C, Pickett J, et al. Von Hippel-Lindau syndrome[J]. Front Horm Res, 2013, 41: 30-49.
[12] Aronow ME, Wiley HE, Gaudric A, et al. Von Hippel-Lindau disease: update on pathogenesis and systemic aspects[J]. Retina, 2019, 39(12): 2243-2253. doi: 10.1097/IAE.0000000000002555
[13] Wiley HE, Krivosic V, Gaudric A, et al. Management of retinal hemangioblastoma in von Hippel-Lindau disease[J]. Retina, 2019, 39(12): 254-2263.
[14] Binderup MLM. von Hippel-Lindau disease: diagnosis and factors influencing disease outcome[J]. Dan Med J, 2018, 65(3): B5461.
[15] Vijapura C, Aldin ES, Capizzano AA, et al. Genetic syndromes associated with central nervous system tumors[J]. Radiographics, 2017, 37(1): 258-280. doi: 10.1148/rg.2017160057
[16] Zubair T, Callaway NF, Ludwig CA, et al. Von Hippel-Lindau syndrome phenotype with prominent vitreoretinal neovascularization treated with early ppv: a case series and literature review[J]. Ophthalmic Surg Lasers Imaging Retina, 2021, 51, (2): 109-115.
[17] Marques A, Portugal R. Ovarian steroid cell tumor in an adolescent with von hippel-lindau syndrome: a case report and review of the literature[J]. Int J Gynecol Pathol, 2020, 39(5): 473-477. doi: 10.1097/PGP.0000000000000628
[18] Keutgen XM, Hammel P, Choyke PL, et al. Evaluation and management of pancreatic lesions in patients with von Hippel-Lindau disease[J]. Nat Rev Clin Oncol, 2016, 1(9): 537-549.
[19] Garnier S, Réguerre Y, Orbach D, et al. Pediatric pheochromocytoma and paraganglioma: an update[J]. Bull Cancer, 2014, 101(10): 966-975. doi: 10.1684/bdc.2014.2031
[20] Maccora D, Walls GV, Sadler GP, et al. Bilateral adrenalectomy: a review of 10 years' experience[J]. Ann R Coll Surg Engl, 2017, 99(2): 119-122. doi: 10.1308/rcsann.2016.0266
[21] Gondek AS, Masip VJ, Muñoz CJ, et al. Adolescent hydrocele carrying a surprise: a case of papillary cysta-denoma of the epididymis[J]. Urology, 2018, 112: 172-175. doi: 10.1016/j.urology.2017.10.040
-
期刊类型引用(1)
1. 郝晋璇,蔺亚斌,齐一洁,李哲,魏红霞,杨喜枫,刘云峰. 冯·希佩尔-林道综合征合并肝细胞核因子1α突变1例. 中华糖尿病杂志. 2024(12): 1418-1422 .  百度学术
百度学术
其他类型引用(0)

计量
- 文章访问数: 246
- HTML全文浏览量: 163
- PDF下载量: 42
- 被引次数: 1


