 作者投稿
作者投稿 专家审稿
专家审稿 编辑办公
编辑办公 主编办公
主编办公
-
摘要:
纯合子家族性高胆固醇血症(HoFH)是一种严重的常染色体遗传代谢性疾病,未经治疗干预的患者常因极高水平低密度脂蛋白胆固醇(LDL-C)导致早发动脉粥样硬化性心血管疾病(ASCVD),往往在30岁前死亡。本团队报道1例复合杂合突变的HoFH患儿,常规饮食控制加降脂药物治疗效果不佳,血脂净化治疗虽然有效,但难以长期维持应用。之后进行同种原位肝移植并进行了2年的随访观察,患儿LDL-C水平长期维持在正常水平,生长发育良好。本病例的报道希望能够提高临床医师对HoFH疾病诊断和综合治疗策略的了解。
-
关键词:
- 纯合子家族性高胆固醇血症 /
- 肝移植术 /
- 低密度脂蛋白胆固醇
Abstract:Homozygous familial hypercholesterolemia (HoFH) is a rare and serious autosomal genetic metabolic disease. Patients without intervention often die younger than 30 years old from early atherosclerotic cardiovascular disease (ASCVD)incurred by extremely high levels of low-density lipoprotein cholesterol (LDL-C). We present a case of HoFH, a child with compound heterozygous mutation in this study. The effect of conventional lipid-lowering therapy through diet control and lipid-lowering drugs was unsatisfactory. The blood-lipid purification proves effective but has poor compliance and difficult to maintain for a longer time. The patient received orthotopic liver transplantation and had been followed for 2 years, with the patient shows normal LDL-C, well growth and development. We hope the case will provide the clinician with better understanding of the diagnosis and treatment of the rare disease of HoFH.
-
罕见病是一类发病率、患病率极低的疾病总称,目前中国对于罕见病尚无统一定义。2018年5月国家卫生健康委员会等部门联合制定的《第一批罕见病目录》公布,首次将121种罕见病收录其中,这是中国政府首次以疾病目录的形式界定罕见病[1]。《中国罕见病定义研究报告2021》更新了罕见病定义,即“新生儿发病率小于1/万、患病率小于1/万、患病人数小于14万的疾病”,使中国的罕见病防治规范化、制度化。虽然罕见病单病种的患病人数少,但中国罕见病的实际患病群体庞大,同时罕见病在临床上确诊难、治疗费用高昂,造成了中国重大疾病经济负担的局面。
心血管疾病是导致中国居民死亡的首要病因,2019年中国农村、城市因心血管病死亡分别占死因的46.74%和44.26%[2-3]。心血管疾病虽为常见病,但在临床实践中易忽略常见症状的罕见病因,导致多数罕见病易误诊。罕见心血管疾病可分为两类:第一类是离子通道病,指离子通道亚基或调节离子通道的蛋白质功能障碍而引起的疾病[4]。这类疾病可分为遗传性和获得性两大类,遗传性的心脏离子通道病主要包括:长QT综合征(long QT syndrome,LQTS)、短QT综合征(short QT syndrome,SQTS)、Brugada综合征(Brugada syndrome,BrS)和儿茶酚胺敏感性多形性室性心动过速(catecholaminergic polymorphic ventri-cular tachycardia,CPVT)等;第二类为特发性心肌病,主要指以遗传性为主(包括混合性)的心肌病,包括特发性或家族性扩张型心肌病(dilated cardiomyopathy,DCM)、致心律失常型右室发育不良/心肌病(arrhythmogenic right ventricular cardiomyopathy,ARVC)、特发性或者家族性限制型心肌病、左室致密化不全(left ventricular noncompaction,LVNC),以及遗传性转甲状腺素蛋白淀粉样变心肌病(transthyretin amyloidosis cardiomyopathy,ATTR-CM)。此外,还有一类罕见病常累及心脏,表现为心功能不全相关症状,如冠状动脉扩张病、法布雷病、Danon病、纯合子家族性高胆固醇血症、特发性肺动脉高压(idiopathic pulmonary arterial hypertension, IPAH)、马方综合征等。多数罕见心血管病属于遗传性疾病,基因突变种类繁多但患病人数相对较少。由于其病因复杂,常需多学科、跨专业的临床医师及医学遗传专家协作才能精准诊断;而且缺少有效的医治手段,患者病死率高。因此亟需利用新技术和平台探究罕见心血管疾病致病机制,提升诊疗水平,助力罕见心血管疾病药物的筛选和研发。本文简要概述罕见心血管疾病诊断和治疗的研究进展,并探讨如何加速罕见病药物研发。
1. 多组学技术在罕见心血管疾病诊疗中的应用
随着基因组学技术的飞速发展,全外显子组测序(whole-exome sequencing,WES)和全基因组测序(whole-genome sequencing,WGS)作为罕见病的辅助筛查手段在临床应用中受到广泛关注。2020年Turro等[5]报道了英国“十万基因组计划”的一项试点研究,通过对约1万例罕见病患者进行WGS,WGS可诊断罕见病基因组的编码和非编码区域的未知突变,加速了罕见病的致病基因识别和病因发现;英国国家医疗服务体系计划将WGS纳入患者的常规医疗保健,以促进罕见病的分子诊断和精准治疗。随着二代测序周期的不断缩短,可快速对新生儿进行疾病的早期诊断和防控。LQTS是一种遗传性心脏离子通道疾病,常表现为心悸、晕厥, 且易发生恶性室性心律失常,甚至造成心源性猝死[6]。斯坦福大学团队对1例二度Ⅰ型房室传导阻滞和室性心律失常的新生儿在出生10日内通过快速WGS,明确了与LQTS相关KCNH2基因的一个已知致病性突变位点和RNF207基因的一个未知突变位点,并根据该分子诊断中受影响的离子通道定制药物治疗[7]。快速测序技术有助于对新生儿开展三级防控,有效降低危害疾病的发生率。
新组学技术的不断突破为罕见心血管疾病致病机制的探究提供了新视角,给临床治疗带来新的前景。DCM是一种在无异常负荷或冠状动脉疾病的情况下,存在左心室或双心室收缩功能障碍伴有心室扩张的心肌疾病;ARVC同样会引发心室功能障碍,常伴有右心室受累、心律失常和纤维脂肪堆积。这两类疾病有明显遗传倾向的心肌受累表现,其致病性变异在心肌细胞中特异性表达,导致患者进展为心力衰竭。Reichart等[8]对DCM和ARVC患者的心脏组织进行单细胞转录组测序,得到这两类心肌病的心室细胞图谱,揭示不同基因型的相关通路、细胞间相互作用和差异基因表达,为心力衰竭治疗提供候选靶点。肌联蛋白(titin)是由TTN基因编码的位于心脏和骨骼肌的大蛋白质,在DCM患者中15%~25%的TTN杂合子变异可导致titin截短变异。宾夕法尼亚大学团队[9]利用无偏蛋白质组学技术,在DCM患者的移植心脏样本中定量检测到截短的titin,提示截短的titin片段损伤心肌细胞导致DCM,为由TTN截短变异引起的DCM靶向疗法研究提供理论基础。既往研究发现肠道菌群可调节血脂成分,肠道微生物群的紊乱会导致高脂血症发生,进而引发心血管疾病。家族性高胆固醇血症(familial hypercholesterolemia,FH)是一种遗传性罕见病,导致低密度脂蛋白胆固醇(low-density lipoprotein cholesterol,LDL-C)水平显著升高,本团队通过全外显子组和宏基因组测序证实了LDLR c.1723C>T引起的错义突变导致FH患者LDL-C代谢异常,同时发现在FH患者口腔中普雷沃氏菌显著升高[10]。
多组学联合人工智能技术可解析罕见心血管疾病的复杂机制,为罕见心血管疾病的个体化诊疗提供更多可能。有团队通过基因组学联合影像组学描述ARVC的遗传-临床-影像-病理特征,在国际上首次建立ARVC的精准分型,根据四种亚型的不同遗传背景和临床特征,提出“精准治疗”策略[11]。Goto等[12]基于卷积神经网络模型,分别以3家和5家医学中心的心电图或超声心动图数据作为训练集和测试集进行建模,构建了ATTR-CM智能诊断模型,该模型可以将心电图预筛查的阳性预测率从33%提高至74%~77%。此外,基于心电图的人工智能辅助诊断系统在肺动脉高压、特发性心肌病、BrS等罕见心血管疾病诊断中表现出优异的诊断效能[13-15]。
2. 诱导性多能干细胞在罕见心血管疾病中的应用
人诱导性多能干细胞(induced pluripotent stem cell,iPSC)分化的细胞可由各种体细胞诱导分化而来,有效获取特定患者的疾病特征和细胞表型,避免了获取人类原代心肌细胞产生的损伤及伦理问题。人iPSC分化的心肌细胞(iPSC-derived cardiac myocytes,iPSC-CM) 与原代心肌细胞模型一样,可以有节律的收缩,并对大多数作用于心脏的药物有类似反应[16]。这些优势使得人iPSC-CM成为适合罕见心血管疾病发病机制及治疗研究的细胞模型。BrS是一种遗传性疾病,主要因编码心肌细胞离子通道NaV1.5的基因产生突变,导致心肌复极时,离子流发生紊乱,从而诱发心室颤动等致命性心律失常的临床综合征,该病死亡率高。2014年,Cerrone等[17]首次报道携带了PKP2突变(c.2484C>T)的iPSC模型。该突变位点导致BrS症状和钠电流密度减少,在细胞外水平通过转染野生型PKP2可修复该缺陷。2016年,Liang等[18]发表携带了两种特异性突变的iPSC-CMs模型,即SCN5A基因的双错义突变(p.R620H和p.R811H)和1个碱基的缺失突变。在该细胞模型中观察到内向钠电流钝化,触发活性增强,以及异常Ca2+运作。通过CRISPR-Cas9基因编辑对患者来源iPSC-CMs中的突变基因进行纠正,可修复触发活性和异常Ca2+瞬变。使用人iPSC-CM模型还可以进行药物筛选和治疗效果的检测,并且已经使用表型或基于靶标的筛选鉴定了潜在治疗某些疾病的候选药物。Yang等[19]基于BAG3基因缺失的扩张型心肌病iPSC-CM模型中,发现HDAC6抑制可以减少肌节损伤,对心脏有保护作用,并在小鼠模型上进行了验证。该研究发现了新的心血管疾病干预靶点,并推动了新型小分子药物TN-301——一种高选择性HDAC6抑制剂的研发。
与传统的二维细胞模型相比,心脏类器官作为新一代药物心脏毒性评估模型和心脏疾病研究模型,有利于深入探索心脏的生理功能和心血管疾病的病理机制,也有利于探索肿瘤治疗等相关药物的心脏毒性。已有学者利用人iPSC构造出titin基因截短突变相关DCM、BRAF基因突变相关肥厚型心肌病及家族性LQTS等疾病的心脏类器官模型[20]。目前,心脏类器官研究相比其他组织类器官尚处于起步阶段,但在转化研究中有巨大的潜力,有助于推动罕见心血管疾病的精准治疗。
3. 基因治疗助力罕见心血管疾病的药物研发
基因治疗是一种将外源的目的基因通过分子生物学技术转运到受体靶细胞或者靶器官,纠正缺陷或异常的基因,达到治疗疾病的目的,可用于肿瘤、遗传病、心血管病等疾病的治疗中。基因载体可分为病毒载体和非病毒载体,目前临床常用的病毒载体有腺病毒、腺相关病毒(adeno-associated virus,AAV)等,转染效率较高。AAV作为基因治疗的一种有力载体被广泛应用,不同血清型感染的细胞类型和效率也各不相同,AAV6型和AAV9型因其对心脏的更高转导效率,已成为需要在心脏中选择性表达的首选AAV血清型[21]。Yu等[22]以多个BrS家族中发现的SCN5A突变(Scn5Ag1746R/+)构建了小鼠疾病模型,并基于该模型测试了AAV9-MOG1基因治疗,发现治疗后逆转了钠通道/电流缺陷,纠正了与BrS相关的心脏电生理异常和临床特征。此外,AAV基因治疗也可用于扩张型心肌病。体内压力超负荷时,miR146a抑制心肌细胞能量代谢,通过AAV9靶向心肌细胞使二氢脂酰琥珀酰转移酶(dihydrolipoamide succinyltransferase,DLST)过表达可减轻心肌肥厚,保护心脏功能,表明DLST和miRNA-146a是DCM的潜在治疗靶标[23]。迄今为止,全球有400多项基因疗法处于临床开发阶段,但AAV应用于罕见心血管疾病临床治疗仍需时日。
CRISPR-Cas技术由于其易于载体构建、靶向位点选择灵活、基因组编辑效率高,已成为潜在的用于基因治疗的新方案。目前在临床水平通过基因组编辑可以预防或治疗两种类型的心血管疾病:第一类是心血管伴有遗传的疾病,如DCM、先天性LQTS及引起心脏功能障碍的肌营养不良等;第二类是马方综合征、家族性肺动脉高压等疾病。转甲状腺素蛋白淀粉样变(transthyretin amyloidosis,ATTR)是一种罕见的系统性、进行性、致死性疾病,其特征是转甲状腺素蛋白(transthyretin,TTR)解离成单体并错误折叠为淀粉样物质沉淀,常累及心脏和肾脏等重要器官,导致功能衰竭。NTLA-2001是一种新的基于CRISPR-Cas9的体内基因编辑疗法,通过静脉输注给药,旨在编辑肝细胞中的TTR以减少其产生。Gillmore等[24]通过纳入6例ATTR患者,并对其中3例进行单次给药NTLA-2001后发现,治疗组相较于对照组患者的血清TTR浓度持续降低,在28 d时达到最低点,同时,治疗组患者发生与药物相关的不良事件仅为轻度。基于基因组编辑技术的基因治疗方案可以从根本上改变基因组信息,但目前仍存在部分基因脱靶效应。非预期基因的永久性突变可能带来严重的副作用,因此该技术在临床上仍处于临床前动物模型中的直接治疗干预和基础研究阶段。
4. 罕见心血管疾病新药研发及用药保障
为了解决罕见病患者无药可治,或境外有药、境内无药的困境,近年来中国大力鼓励和推进罕见病原创性药物、仿制药物及罕见病药物相关医疗器械的研发。新药开发具有耗时长、成本高的特点,与开发新药相比,老药新用风险较少、成本较低、周期较短,其为罕见心血管疾病的治疗提供了新契机。许多经典药物在罕见病治疗中焕发新春,例如,化疗药雷帕霉素及其类似物可用于儿童结节性硬化症合并癫痫的治疗;在神经系统罕见病中,脑保护剂依达拉奉可用于肌萎缩侧索硬化的治疗。近年来iPSC模型和基因编辑技术日益成熟,多种罕见心血管疾病细胞模型的建立及高通量筛药技术的发展为老药新用助力添翼,成为罕见病用药的新捷径。
2018年以来,中国先后发布三批《临床急需境外新药名单》,其中包含40余种罕见病药物[25]。通过进一步简化名单上罕见病药物的上市流程,缩短其上市时间,极大程度地解决了罕见病患者用药难的问题。自《第一批罕见病目录》发布以来,共有6种罕见心血管治疗药物获批上市,包括治疗法布雷病的阿加糖酶α和β;治疗ATTR-CM的氯苯唑酸软胶囊;治疗纯合子家族性高胆固醇血症的依洛尤单抗;治疗IPAH的枸橼酸西地那非和曲前列尼尔。在医疗保障方面,中国已有的罕见病适用证药物不到90种,其中58种已经纳入到国家基本医保目录,覆盖29种罕见病,基本解决罕见病患者用药贵的难题。
5. 展望
罕见心血管疾病相关研究虽已取得巨大突破,但转化至临床用药仍任重而道远。随着新技术的不断发展,将多组学数据充分整合,可加快基础研究创新成果的转化。罕见心血管疾病的药物研发仍处于研究初期,但发现的特定药物靶点已进入临床试验中,加速罕见病药物研发,有望惠及更多患者,切实提高罕见病诊疗水平。
作者贡献:文章撰写与研究设计:陈沛沛、田庄、张抒扬;数据收集与分析:陈沛沛、田庄、陈伟、马明圣、刘鑫、秦岩、徐海峰、朱志军;文章审阅与修改:陈沛沛、田庄、张抒扬。利益冲突:所有作者均声明不存在利益冲突。 -
![]()
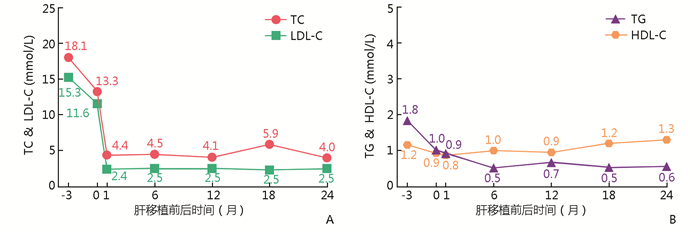
图 1 患儿口服降脂药物及血清胆固醇水平变化
Figure 1. Changes of oral lipid-lowering drugs and serum cholesterol level in this patient
![]()
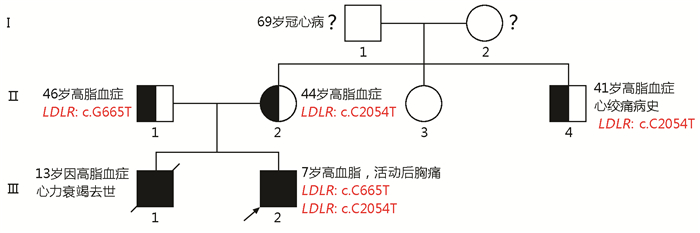
图 2 患者家系图谱
□为男性正常者;○为女性正常者,

Figure 2. The pedigree of the patient
![]()
图 3 患儿多发皮肤黄色瘤
患儿臀部、后足跟、手指背侧及膝关节处均可见到皮肤黄色瘤(红色箭头所示)
Figure 3. The multiple skin xanthomas in the patient
-
[1] Blom DJ, Harada-Shiba M, Rubba P, et al. Efficacy and safety of alirocumab in adults with homozygous familial hypercholesterolemia: the ODYSSEY HoFH trial[J]. J Am Coll Cardiol, 2020, 76(2): 131-142. doi: 10.1016/j.jacc.2020.05.027
[2] Cuchel M, Bloedon LT, Szapary PO, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia[J]. N Engl J Med, 2007, 356(2): 148-156. doi: 10.1056/NEJMoa061189
[3] Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial[J]. Lancet, 2010, 375(9719): 998-1006. doi: 10.1016/S0140-6736(10)60284-X
[4] Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society[J]. Eur Heart J, 2013, 34(45): 3478-90a. doi: 10.1093/eurheartj/eht273
[5] Sharifi M, Futema M, Nair D, et al. Genetic architecture of familial hypercholesterolaemia[J]. Curr Cardiol Rep, 2017, 19(5): 44. doi: 10.1007/s11886-017-0848-8
[6] Chen P, Chen X, Zhang S. Current status of familial hypercholesterolemia in China: a need for patient FH registry systems[J]. Front Physiol, 2019, 10: 280. doi: 10.3389/fphys.2019.00280
[7] Harada-Shiba M, Arai H, Ishigaki Y, et al. Guidelines for diagnosis and treatment of familial hypercholesterolemia 2017[J]. J Atheroscler Thromb, 2018, 25(8): 751-770. doi: 10.5551/jat.CR003
[8] Sjouke B, Kusters DM, Kindt I, et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome[J]. Eur Heart J, 2015, 36(9): 560-565. doi: 10.1093/eurheartj/ehu058
[9] Gidding SS, Champagne MA, de Ferranti SD, et al. The agenda for familial hypercholesterolemia: a scientific statement from the American Heart Association[J]. Circulation, 2015, 132(22): 2167-2192. doi: 10.1161/CIR.0000000000000297
[10] Naoumova RP, Thompson GR, Soutar AK. Current management of severe homozygous hypercholesterolaemias[J]. Curr Opin Lipidol, 2004, 15(4): 413-422. doi: 10.1097/01.mol.0000137222.23784.2a
[11] Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society[J]. Eur Heart J, 2014, 35(32): 2146-2157. doi: 10.1093/eurheartj/ehu274
[12] Shi Z, Yuan B, Zhao D, et al. Familial hypercholes-terolemia in China: prevalence and evidence of underdetection and undertreatment in a community population[J]. Int J Cardiol, 2014, 174(3): 834-836. doi: 10.1016/j.ijcard.2014.04.165
[13] 国家卫生健康委办公厅. 关于印发罕见病诊疗指南(2019年版)的通知[EB/OL]. (2019-02-27)[2022-11-20]. http://www.nhc.gov.cn/yzygj/s7659/201902/61d06b4916c348e0810ce1fceb844333shtml. [14] Gagné C, Gaudet D, Bruckert E. Efficacy and safety of ezetimibe coadministered with atorvastatin or simvastatin in patients with homozygous familial hypercholesterolemia[J]. Circulation, 2002, 105(21): 2469-2475. doi: 10.1161/01.CIR.0000018744.58460.62
[15] Raal FJ, Honarpour N, Blom DJ, et al. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial[J]. Lancet, 2015, 385(9965): 341-350. doi: 10.1016/S0140-6736(14)61374-X
[16] Raal FJ, Hovingh GK, Blom D, et al. Long-term treatment with evolocumab added to conventional drug therapy, with or without apheresis, in patients with homozygous familial hypercholesterolaemia: an interim subset analysis of the open-label TAUSSIG study[J]. Lancet Diabetes Endocrinol, 2017, 5(4): 280-290. doi: 10.1016/S2213-8587(17)30044-X
[17] Stein EA, Honarpour N, Wasserman SM, et al. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia[J]. Circulation, 2013, 128(19): 2113-2120. doi: 10.1161/CIRCULATIONAHA.113.004678
[18] Wang A, Richhariya A, Gandra SR, et al. Systematic review of low-density lipoprotein cholesterol apheresis for the treatment of familial hypercholesterolemia[J]. J Am Heart Assoc, 2016, 5(7): e003294. doi: 10.1161/JAHA.116.003294
[19] Sampietro T, Sbrana F, Bigazzi F, et al. The incidence of cardiovascular events is largely reduced in patients with maximally tolerated drug therapy and lipoprotein apheresis. A single-center experience[J]. Atheroscler Suppl, 2015, 18: 268-272. doi: 10.1016/j.atherosclerosissup.2015.02.040
[20] Martinez M, Brodlie S, Griesemer A, et al. Effects of liver transplantation on lipids and cardiovascular disease in children with homozygous familial hypercholesterolemia[J]. Am J Cardiol, 2016, 118(4): 504-510. doi: 10.1016/j.amjcard.2016.05.042
[21] Mansoorian M, Kazemi K, Nikeghbalian S, et al. Liver transplantation as a definitive treatment for familial hypercholesterolemia: a series of 36 cases[J]. Pediatr Transplant, 2015, 19(6): 605-611. doi: 10.1111/petr.12562
[22] Ng VL, Alonso EM, Bucuvalas JC, et al. Health status of children alive 10 years after pediatric liver transplantation performed in the US and Canada: report of the studies of pediatric liver transplantation experience[J]. J Pediatr, 2012, 160(5): 820-826. e3. doi: 10.1016/j.jpeds.2011.10.038
[23] Kelly DA, Bucuvalas JC, Alonso EM, et al. Long-term medical management of the pediatric patient after liver transplantation: 2013 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation[J]. Liver Transpl, 2013, 19(8): 798-825. doi: 10.1002/lt.23697
[24] Ishigaki Y, Kawagishi N, Hasegawa Y, et al. Liver transplantation for homozygous familial hypercholesterolemia[J]. J Atheroscler Thromb, 2019, 26(2): 121-127. doi: 10.5551/jat.RV17029
[25] Ibrahim M, El-Hamamsy I, Barbir M, et al. Translational lessons from a case of combined heart and liver transplantation for familial hypercholesterolemia 20 years post-operatively[J]. J Cardiovasc Transl Res, 2012, 5(3): 351-358. doi: 10.1007/s12265-011-9311-1
[26] Cephus CE, Qureshi AM, Sexson Tejtel SK, et al. Coronary artery disease in a child with homozygous familial hypercholesterolemia: regression after liver transplantation[J]. J Clin Lipidol, 2019, 13(6): 880-886. doi: 10.1016/j.jacl.2019.09.007
[27] Alim A, Tokat Y, Erdogan Y, et al. Liver transplantation for homozygote familial hypercholesterolemia: the only curative treatment[J]. Pediatr Transplant, 2016, 20(8): 1060-1064. doi: 10.1111/petr.12763
[28] Greco M, Robinson JD, Eltayeb O, et al. Progressive aortic stenosis in homozygous familial hypercholesterolemia after liver transplant[J]. Pediatrics, 2016, 138(5): e20160740. doi: 10.1542/peds.2016-0740
[29] El-Rassi I, Chehab G, Saliba Z, et al. Fatal cardiac atherosclerosis in a child 10 years after liver transplantation: a case report and a review[J]. J Clin Lipidol, 2011, 5(4): 329-332. doi: 10.1016/j.jacl.2011.05.002
[30] Palacio CH, Harring TR, Nguyen NT, et al. Homozygous familial hypercholesterolemia: case series and review of the literature[J]. Case Rep Transplant, 2011, 2011: 154908.
[31] Shrotri M, Fernando BS, Sudhindran S, et al. Long-term outcome of liver transplantation for familial hypercholesterolemia[J]. Transplant Proc, 2003, 35(1): 381-382. doi: 10.1016/S0041-1345(02)03910-6
[32] Popescu I, Simionescu M, Tulbure D, et al. Homozygous familial hypercholesterolemia: specific indication for domino liver transplantation[J]. Transplantation, 2003, 76(9): 1345-1350. doi: 10.1097/01.TP.0000093996.96158.44
[33] Malatack JJ. Liver transplantation as treatment for familial homozygous hypercholesterolemia: too early or too late[J]. Pediatr Transplant, 2011, 15(2): 123-125.
[34] Al-Ashwal A, Alnouri F, Sabbour H, et al. Identification and treatment of patients with homozygous familial hypercholesterolaemia: information and recommendations from a middle east advisory panel[J]. Curr Vasc Pharmacol, 2015, 13(6): 759-770. doi: 10.2174/1570161113666150827125040
[35] Akdur A, Kirnap M, Ayvazoglu Soy EH, et al. Unusual indications for a liver transplant: a single-center experience[J]. Exp Clin Transplant, 2017, 15(Suppl 1): 128-132.
-
期刊类型引用(1)
1. 赵韧,周碧蓉,李泉,王柘,李小虎. PACS辅助PBL教学模式在心血管专科医生心血管罕见病教学中的探索. 蚌埠医学院学报. 2023(10): 1465-1468 .  百度学术
百度学术
其他类型引用(0)
 下载:
下载:


计量
- 文章访问数: 237
- HTML全文浏览量: 109
- PDF下载量: 66
- 被引次数: 1


