 作者投稿
作者投稿 专家审稿
专家审稿 编辑办公
编辑办公 主编办公
主编办公
-
摘要:目的
Mohr-Tranebjaerg综合征(MTS)典型症状包括耳聋、肌张力障碍等,本病非常罕见,遵循X连锁遗传模式。本研究对一个罕见MTS家系进行遗传学分析。
方法收集罕见MTS家系临床资料,同时对先证者Ⅲ6进行全外显子组测序检测,对家族中其他患病个体Ⅲ5和健康人Ⅰ1、Ⅰ2、Ⅱ1、Ⅱ5、Ⅱ7、Ⅱ8、Ⅲ7进行家系验证,将基因检测结果与临床表现反复核实比对。
结果家族中患者表现为较早期出现的耳聋, 多个患病成员随着年龄增长合并肌张力障碍、精神情绪异常。全外显子组测序检测提示先证者Ⅲ6 TIMM8A (NM_004085.3,又称DDP1)基因内含子存在c.133-2delA变异,目前评级为可能致病(LP)。同时在患病个体Ⅲ5及健康女性Ⅰ1、Ⅱ5、Ⅱ7检出此变异。
结论TIMM8A检出变异所致MTS与家系患病者临床表现高度吻合,符合家系共分离,是这个罕见MTS患病家族的分子病因。除耳聋外,其他症状在家族中不同个体中可存在差异。针对此类X连锁疾病的基因诊断,还可以明确女性杂合子,给予遗传咨询和生育指导,从而帮助家族成员制订科学的家庭计划。
-
关键词:
- Mohr-Tranebjaerg综合征 /
- TIMM8A基因 /
- 全外显子组测序
Abstract:ObjectiveMohr-Tranebjaerg syndrome (MTS) is a rare X-linked neurodegenerative disorder which usually involving hearing impairment, gradual dystonia, and other symptoms. In this study, we perform analyzed the genetic makeup of a family with this rare Mohr-Tranebjaerg syndrome.
MethodsWe collected the clinical data of the family, did the whole exome sequencing on the proband Ⅲ6 with a rare mutation, and verified the mutation in another affected family member Ⅲ5 and unaffected members Ⅰ1, Ⅰ2, Ⅱ1, Ⅱ5, Ⅱ7, Ⅱ8, Ⅲ7.
ResultsThe patients in the family all showed early-onset deafness. More than a couple of affected male members have dystonia with/without mental disorders. Genetic testing results showed the proband Ⅲ6 had a c.133-2delA in TIMM8A (NM_ 004085.3, DDP1), highly likely pathogenic(LP). This variation was detected in affected Ⅲ5 as well as the unaffected females Ⅰ1, Ⅱ5, Ⅱ7.
ConclusionsMTS caused by the rare TIMM8A mutation, the molecular etiology of the family with this rare disease, is highly consistent with the clinical manifestations and segregation. Other than the deafness, other symptoms varied among the affected family members. Genetic diagnosis for such X-linked diseases can also identify female heterozygotes. Genetic and reproduction counseling can help families in the family planning.
-
Keywords:
- Mohr-Tranebjaerg syndrome /
- TIMM8A gene /
- whole exome sequencing
-
Mohr-Tranebjaerg综合征(Mohr-Tranebjaerg syndrome,MTS),又称为耳聋肌张力障碍综合征(deafness-dystonia syndrome)或DDON综合征(deafness-dystonia-optic neuronopathy syndrome, OMIM# 304700),1960年在一个挪威家系中被首次报道,最初界定为非综合征X连锁性耳聋(又作DFN-1)[1]。1995—1996年,Tranebjaerg医生团队对该家系家族史、临床表现重新评估,并进行连锁分析,确定该病累及神经、视觉及精神行为,紧接着其致病基因被揭示[2-3]。多数患者可检出TIMM8A基因致病变异或染色体异常累及TIMM8A基因(translocase of inner mitochondrial membrane 8A)。此基因位于X染色体长臂2区2带1亚带(Xq22.1),含有2个外显子和1个内含子,编码一条含有97个氨基酸的肽链,主要参与线粒体内膜蛋白的转运和组装过程。致病机制多为TIMM8A蛋白在线粒体内功能缺陷,从而无法形成TIMM8A/TIMM13复合物,影响其他蛋白在线粒体内正常运送[4]。这种综合征型耳聋非常罕见,目前报道确诊病例全球范围内不超过70例,亚洲更为罕见,目前文献中仅日本报道7例,2019年中国报道3例,此3例主要表现为耳聋,其他系统症状暂未出现[5]。综上,中国MTS极为罕见,临床医师、分子诊断和遗传咨询团队对该患者的临床和基因变异需进一步学习和了解。本课题组对一个MTS综合征家系的临床表现、送检策略和结果分析进行报道,通过示例遗传学分析推进该罕见病的识别、诊断、遗传咨询及生育指导。
1. 资料与方法
1.1 一般资料
Ⅲ7,健康女性,26岁,于2020年10月22日因耳聋家族史和婚育目的就诊于北京协和医院耳聋遗传咨询门诊。了解到家族中多名男性患者有相似症状,详细绘制家系图谱并了解临床症候群,见图 1。
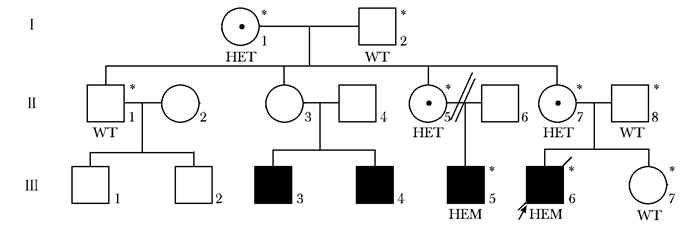
![]() 图 1 Mohr-Tranebjaerg综合征(MTS)家系图□表示健康男性,○表示健康女性,■表示男性患者,●表示女性患者,WT表示野生型,HET表示杂合子,HEM表示半合子,↗表示先证者,*表示受试者Figure 1. Pedigree of this family with Mohr-Tranebjaerg syndrome(MTS)
图 1 Mohr-Tranebjaerg综合征(MTS)家系图□表示健康男性,○表示健康女性,■表示男性患者,●表示女性患者,WT表示野生型,HET表示杂合子,HEM表示半合子,↗表示先证者,*表示受试者Figure 1. Pedigree of this family with Mohr-Tranebjaerg syndrome(MTS)病例1,Ⅲ6,先证者,男性,30岁,自幼听力差,聋哑状态,25岁出现淡漠、抑郁表现及严重肌张力障碍,重症监护住院史,2021年因肌张力障碍、感染、营养不良合并多器官衰竭去世。病例2,Ⅲ3,男性,31岁,自幼耳聋,助听,20岁左右有淡漠、抑郁表现,规范治疗,后出现肌张力障碍,症状较轻。病例3,Ⅲ4,男性,15岁,自幼耳聋,助听,暂无其他明显临床表现。病例4,Ⅲ5,男性,25岁,自幼耳聋,已助听,12岁出现进展性肌张力障碍,在家族患病者中发病最早,症状较重。家族中其他成员无类似症状及明显异常。本研究经北京协和医院医学伦理委员会审查批准(伦理审查编号:ZS-1738)。
1.2 方法
签署知情同意书后,抽取受试者静脉血2 mL。
1.2.1 先证者行全外显子检测
使用MagPure Buffy Coat DNA Midi KF Kit标准流程提取DNA,质控DNA浓度和纯度合格,使用片段化酶(Segmentase,华大基因)片段化,构建文库,文库经Agilent 2100 Bioanalyzer和BMG进行片段大小、浓度检测,合格后将需要不同数据量的文库进行pooling、定量;然后将pooling文库进行单链环化,高通量测序仪MGISEQ-2000进行测序。对下机的原始数据(raw reads)进行测序质量评估后根据不同数据库进行比对、注释和筛选。
1.2.2 一代测序行家系验证
对于所有发现的致病突变,包括后续家系验证,均在其所在片段上下游设计引物。进行PCR扩增,对其产物做Sanger测序。
2. 结果
2.1 临床表现分析
从家系图和临床表现来看,家族中4例患者,均为男性,年龄分别为15岁、25岁、30岁、31岁。4例均表现出自幼听力损失(4/4,100%),程度较重,其中3例助听,可交流,1例未助听,聋哑状态;3例表现为肌张力障碍(3/4,75%),发病时间10余岁至20余岁不等,吞咽障碍明显,症状轻重不等,轻者可维持日常生活,重者需长期住院维持治疗;2例有精神情绪症状(2/4,50%),主要表现为淡漠、抑郁。4例患者均不伴视觉、智力异常。
2.2 送检策略及基因检测结果
首次就诊者为Ⅲ7,为26岁健康女性,其就诊咨询目的明确,结合患病家族史及个人婚育需求,明确家族患者患病分子病因和评估规避其生育类似患者的风险。经遗传咨询后,送检策略为送检患病一兄Ⅲ6,行临床全外显子检测,同时采集到家族成员Ⅰ1、Ⅰ2、Ⅱ1、Ⅱ5、Ⅱ7、Ⅱ8、Ⅲ5和Ⅲ7行家系验证。
在男性患者Ⅲ5和Ⅲ6检出TIMM8A基因c.133-2delA半合子变异;在女性健康成员Ⅰ1、Ⅱ5、Ⅱ7检出TIMM8A基因c.133-2delA杂合子变异;在家族中其他成员Ⅰ2、Ⅱ1、Ⅱ8和Ⅲ7未检出此变异(图 2)。根据美国医学遗传学与基因组学会(American College of Medical Genetics and Genomics,ACMG) 指南,目前将此变异评级为疑似致病(likely pathogenic,LP)。证据包括:PVS1_Strong:影响剪切,此区域变异罕见且与产物相关;PM2:ESP数据库、千人数据库、EXAC数据库中正常对照人群中未发现的;PP1:突变与疾病在家系中共分离(在家系多个患者中检测到此变异);PP4:变异携带者的表型或家族史高度符合某种单基因遗传疾病。综上,目前评级为:PVS1_Strong+PM2+PP1+PP4=LP。
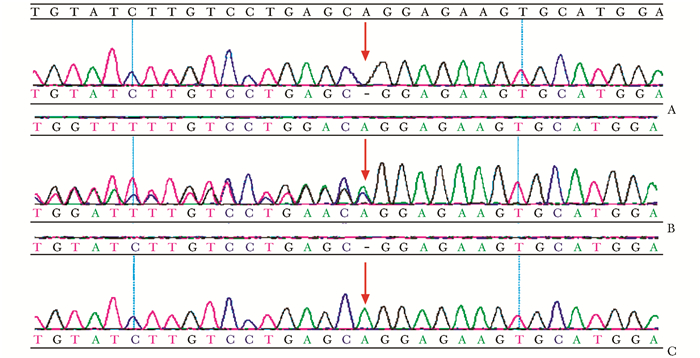
![]() 图 2 MTS家系TIMM8A基因测序A.男性患者Ⅲ5和Ⅲ6检出TIMM8A基因c.133-2delA半合子变异;B.女性健康成员Ⅰ1、Ⅱ5、Ⅱ7检出TIMM8A基因c.133-2delA杂合子变异;C.家族中其他成员Ⅰ2、Ⅱ1、Ⅱ8和Ⅲ7未检出此变异;红色箭头所指为变异位置Figure 2. Sanger sequencing of MTS family with TIMM8A variation
图 2 MTS家系TIMM8A基因测序A.男性患者Ⅲ5和Ⅲ6检出TIMM8A基因c.133-2delA半合子变异;B.女性健康成员Ⅰ1、Ⅱ5、Ⅱ7检出TIMM8A基因c.133-2delA杂合子变异;C.家族中其他成员Ⅰ2、Ⅱ1、Ⅱ8和Ⅲ7未检出此变异;红色箭头所指为变异位置Figure 2. Sanger sequencing of MTS family with TIMM8A variation3. 讨论
家族中4例男性患者表现为婴幼儿期双耳听力障碍,影响言语发育,需要助听,多名伴有典型的全身肌张力障碍,符合MTS典型临床特点。遗传模式分析得出该家系符合X连锁遗传规律。送检策略为先证者行临床全外显子检测,家系验证采血受试者重点要包括先证者直系亲属、家族中其他男性患者、男性患者的母系家族成员,尤其关注Ⅱ1作为此X连锁家族唯一无症状的男性后代,其基因型对于甄别、验证分子病因尤为重要。基因检测男性患者Ⅲ5和Ⅲ6检出TIMM8A基因c.133-2delA半合子变异,该变异极为罕见,尚未被人类基因突变数据库(human gene mutation database,HGMD)收录,ACMG评级为疑似致病(LP)且符合家系共分离,综上,此罕见家系可以确诊为MTS。
MTS又称为DDON综合征,多发生于婴幼儿时期,症状可能包括:语前或语后发生的感音神经性听力损失;10余岁发生可缓慢进展的肌张力障碍(如口下颌肌张力障碍导致吞咽困难)或共济失调多发病;20岁左右可能出现视神经萎缩导致视力进行性受累,40岁左右可出现痴呆;还可能在儿童期出现性格异常、抑郁偏执等精神症状,可呈进展性加重。其中听力损失可以表现为稳定型或渐进性下降,而神经肌肉、视觉、神经精神病学表现轻重不一,进展速度个体化差异也较大。目前该病的诊断依据除临床表现外,倚重基因检测。男性先证者检出TIMM8A基因上致病变异的半合子状态或女性先证者检出TIMM8A基因上致病变异的杂合子状态;或者染色体Xp22.1存在变异累及TIMM8A基因即可明确诊断。
MTS极为罕见,目前已报道的致病变异约15种[6]。其中,目前为HGMD收录的TIMM8A基因剪切突变仅2种[7-10],见表 1。此次检出TIMM8A基因c.133-2delA变异为极为罕见的剪切变异,HGMD尚未收录。中国学者曾于2013年报道过一个家系携带同样的罕见变异,反转录PCR提示该变异直接导致TIMM8A mRNA剪切异常[11]。此次在家系中再次检出补充了此基因罕见影响剪切变异型突变谱系,继中国学者2013年、2019年报道MTS后[5, 11]再次报道MTS罕见病病例家系。
表 1 TIMM8A基因剪切变异文献回顾Table 1. Review of TIMM8A gene splicing variation in the literature基因 位点 临床表现 报道年份(年) HGMD 参考文献 TIMM8A c.133-23A>C 累及1个家系中2例男性患者,其中1例为31岁男性患者自4岁起患有感音神经性耳聋渐进性至极重度耳聋,11岁开始出现渐进性肌张力障碍,24岁发现视觉异常,另1例为29岁男性患者,11岁诊断为双侧听力损失,20岁出现肌张力障碍和精神情绪症状 2005(是TIMM8A基因报道的首个剪切突变) 已收录 [7] TIMM8A c.132+1G>A 1例42岁男性患者检出c.132+1G>A变异,临床表现为8月龄发现的生儿听力损失,25岁出现的肌张力障碍,30岁开始表现有视觉受损 2007 已收录 [8] TIMM8A c.132+1G>A 1个散发病例检出c.132+1G>A变异。患者为男性,31岁,4岁因听力损失助听,14岁发现双侧视神经萎缩,23岁出现渐进性肌张力障碍 2008 已收录 [9] TIMM8A c.133-2delA 1个家族中有4例男性患者均检出c.133-2delA变异,表现为2岁发现听力差伴言语欠清,10余岁出现肌力障碍伴姿势异常,其中两例伴有精神情绪异常,1例伴智力水平降低。其中1例行听力检测提示重度感音神经性耳聋。 2013 未收录 [11] TIMM8A c.132+1G>A 在1例散发男性肌张力障碍病例第3次检出c.132+1G>A变异,表现为2岁出现的肌张力障碍,未提及该患者其他临床表型 2018 已收录 [10] HGMD: 人类基因突变数据库 目前MTS临床诊治包括以下多个方面:①听神经病是导致听力损失的真实病因,故助听效果一般很难达到非常满意;②患者教育、药物治疗、康复治疗以期提高运动能力,避免挛缩,给予合适的装置帮助日常生活;③同时要关注行为学、神经精神病学方面的异常。规律随访包括神经肌肉学、眼科、听力学、痴呆及神经精神病学定期评估,也包括心理咨询、康复、定期的随访。
MTS遗传咨询要点:该病遵循X连锁遗传模式。先证者的母亲往往携带TIMM8A基因上致病变异或染色体Xp22.1存在变异累及TIMM8A基因,每次生育都有1/2的概率将此变异遗传至后代。获得此致病变异的男性后代将表现为MTS;获得此变异的女性后代作为杂合子可能无症状或有较轻临床表现,如轻度听力异常和病灶性肌张力异常[11-12]。男性患者会将此变异遗传给他的女儿,但男性子代不会从患病父亲遗传到此致病变异。在先证者致病变异明确的情况下,可考虑产前基因诊断或胚胎移植前基因诊断,以协助科学制订家庭计划。对于文中所涉及家系中首次寻求遗传咨询帮助的女性健康成员Ⅲ7,因其未携带有TIMM8A基因c.133-2delA剪切变异,故生育MTS男性后代可能性小,不涉及TIMM8A基因相关的产前诊断或筛选。在其有明确优生优育意愿的情况下,建议Ⅲ7及其配偶考虑以中国常见遗传病为检测范围的携带者筛查,避免其他遗传风险对后代的影响。
综上,MTS是极为罕见的X连锁耳聋-肌张力障碍综合征。临床上遇到男性患者存在以下症状需甄别考虑是否患有MTS:语前或语后发生的感音神经性耳聋,可表现为听神经谱系障碍;运动能力异常(肌张力异常/共济失调/吞咽困难);逐渐发作缓慢进展的个性变化,偏执,痴呆;视神经萎缩所致视觉灵敏度逐渐下降;家族史提示X连锁遗传模式。同时应为患者安排包括TIMM8A基因的基因检测协助诊断。对于有明确分子病因的患者及其家族成员,要进行遗传咨询和生育指导。
作者贡献:高儒真负责资料收集、文献检索、文章撰写和修返稿件;樊悦、范欣淼、杨腾裕、宋雯洁负责资料收集及文献检索;陈晓巍负责确定主题及审核修订稿件。利益冲突:所有作者均声明不存在利益冲突。 -
![]()
图 1 Mohr-Tranebjaerg综合征(MTS)家系图
□表示健康男性,○表示健康女性,■表示男性患者,●表示女性患者,WT表示野生型,HET表示杂合子,HEM表示半合子,↗表示先证者,*表示受试者
Figure 1. Pedigree of this family with Mohr-Tranebjaerg syndrome(MTS)
![]()
图 2 MTS家系TIMM8A基因测序
A.男性患者Ⅲ5和Ⅲ6检出TIMM8A基因c.133-2delA半合子变异;B.女性健康成员Ⅰ1、Ⅱ5、Ⅱ7检出TIMM8A基因c.133-2delA杂合子变异;C.家族中其他成员Ⅰ2、Ⅱ1、Ⅱ8和Ⅲ7未检出此变异;红色箭头所指为变异位置
Figure 2. Sanger sequencing of MTS family with TIMM8A variation
表 1 TIMM8A基因剪切变异文献回顾
Table 1 Review of TIMM8A gene splicing variation in the literature
基因 位点 临床表现 报道年份(年) HGMD 参考文献 TIMM8A c.133-23A>C 累及1个家系中2例男性患者,其中1例为31岁男性患者自4岁起患有感音神经性耳聋渐进性至极重度耳聋,11岁开始出现渐进性肌张力障碍,24岁发现视觉异常,另1例为29岁男性患者,11岁诊断为双侧听力损失,20岁出现肌张力障碍和精神情绪症状 2005(是TIMM8A基因报道的首个剪切突变) 已收录 [7] TIMM8A c.132+1G>A 1例42岁男性患者检出c.132+1G>A变异,临床表现为8月龄发现的生儿听力损失,25岁出现的肌张力障碍,30岁开始表现有视觉受损 2007 已收录 [8] TIMM8A c.132+1G>A 1个散发病例检出c.132+1G>A变异。患者为男性,31岁,4岁因听力损失助听,14岁发现双侧视神经萎缩,23岁出现渐进性肌张力障碍 2008 已收录 [9] TIMM8A c.133-2delA 1个家族中有4例男性患者均检出c.133-2delA变异,表现为2岁发现听力差伴言语欠清,10余岁出现肌力障碍伴姿势异常,其中两例伴有精神情绪异常,1例伴智力水平降低。其中1例行听力检测提示重度感音神经性耳聋。 2013 未收录 [11] TIMM8A c.132+1G>A 在1例散发男性肌张力障碍病例第3次检出c.132+1G>A变异,表现为2岁出现的肌张力障碍,未提及该患者其他临床表型 2018 已收录 [10] HGMD: 人类基因突变数据库  下载: 导出CSV
下载: 导出CSV
-
[1] Mohr J, Mageroy K. Sex-linked deafness of a possibly new type[J]. Acta Genet Stat Med, 1960, 10: 54-62.
[2] Tranebjaerg L, Schwartz C, Eriksen H, et al. A new X linked recessive deafness syndrome with blindness, dystonia, fractures, and mental deficiency is linked to Xq22[J]. J Med Genet, 1995, 32(4): 257-263. doi: 10.1136/jmg.32.4.257
[3] Jin H, May M, Tranebjaerg L, et al. A novel X-linked gene, DDP, shows mutations in families with deafness (DFN-1), dystonia, mental deficiency and blindness[J]. Nat Genet, 1996, 14(2): 177-180. doi: 10.1038/ng1096-177
[4] Roesch K, Curran SP, Tranebjaerg L, et al. Human deafness dystonia syndrome is caused by a defect in assembly of the DDP1/TIMM8a-TIMM13 complex[J]. Hum Mol Genet, 2002, 11(5): 477-486. doi: 10.1093/hmg/11.5.477
[5] Wang H, Wang L, Yang J, et al. Phenotype prediction of Mohr-Tranebjaerg syndrome (MTS) by genetic analysis and initial auditory neuropathy[J]. BMC Med Genet, 2019, 20(1): 11. doi: 10.1186/s12881-018-0741-3
[6] Neighbors A, Moss T, Holloway L, et al. Functional analysis of a novel mutation in the TIMM8A gene that causes deafness-dystonia-optic neuronopathy syndrome[J]. Mol Genet Genomic Med, 2020, 8(3): e1121.
[7] Ezquerra M, Campdelacreu J, Muñoz E, et al. A novel intronic mutation in the DDP1 gene in a family with X-linked dystonia-deafness syndrome[J]. Arch Neurol, 2005, 62(2): 306-308. doi: 10.1001/archneur.62.2.306
[8] Kim HT, Edwards MJ, Tyson J, et al. Blepharospasm and limb dystonia caused by Mohr-Tranebjaerg syndrome with a novel splice-site mutation in the deafness/dystonia peptide gene[J]. Mov Disord, 2007, 22(9): 1328-1331. doi: 10.1002/mds.21351
[9] Aguirre LA, Pérez-Bas M, Villamar M, et al. A Spanish sporadic case of deafness-dystonia (Mohr-Tranebjaerg) syndrome with a novel mutation in the gene encoding TIMM8a, a component of the mitochondrial protein translocase complexes[J]. Neuromuscul Disord, 2008, 18(12): 979-981. doi: 10.1016/j.nmd.2008.09.009
[10] Montaut S, Tranchant C, Drouot N, et al. Assessment of a targeted gene panel for identification of genes associated with movement disorders[J]. JAMA Neurol, 2018, 75(10): 1234-1245. doi: 10.1001/jamaneurol.2018.1478
[11] 王璐, 张巍, 刘玉和, 等. 线粒体内膜易位酶8A基因新突变导致耳聋肌张力障碍综合征一家系[J]. 中华神经科杂志, 2013, 46(4): 243-246. doi: 10.3760/cma.j.issn.1006-7876.2013.04.007 [12] Swerdlow RH, Wooten GF. A novel deafness/dystonia peptide gene mutation that causes dystonia in female carriers of Mohr-Tranebjaerg syndrome[J]. Ann Neurol, 2001, 50(4): 537-540. doi: 10.1002/ana.1160
计量
- 文章访问数: 304
- HTML全文浏览量: 179
- PDF下载量: 84


