 作者投稿
作者投稿 专家审稿
专家审稿 编辑办公
编辑办公 主编办公
主编办公
-
摘要:
家族性高胆固醇血症(FH)是一组以LDL代谢异常为主要特征的常染色体(共)显性遗传疾病,是儿童期常见的遗传性疾病之一,也是脂质代谢疾病中较为严重的一种,可导致各种危及生命的心血管疾病及其并发症。近年来,随着国内外对该疾病认识的深入及新型降脂药物的研发,FH的治疗策略愈发多样化。但目前FH的知晓率和诊断率均较低,治疗状况较差,为了加强社会对该疾病的认识,本文对FH的流行病学、诊断与筛查、治疗予以总结。
Abstract:Familial hypercholesterolemia (FH) is a group of autosomal co-dominant genetic diseases mainly characterized by abnormal low-density lipoprotein related metabolism. It is one of the most common inherited diseases in children and one of the most serious lipid metabolism diseases which results in various life-threatening cardiovascular diseases and the complications. In recent years, the treatment protocols for FH have diversified thanks to the deeper understanding of the disease in China and abroad and the development of new lipid-lowering drugs. However, the current awareness and diagnosis rate of FH are very low. The treatment of the disease is much inadequate. This paper summarizes the clinical characteristics, diagnosis, screening strategy, and treatment of FH hoping to enhance the understanding and awareness of the disease in the society.
-

张抒扬
北京协和医院 院长
《罕见病研究》 主编
律回春渐,新元肇启。跨过不平凡的2022,岁月又勾勒出新的年轮。值此2023年新春来临之际,谨向支持和关心我刊发展的编委、专家、作者和读者们致以衷心的感谢和诚挚的祝福!
回眸2022年,《罕见病研究》正式创刊出版。一年来,在各级领导的鼎力支持和编委专家们的悉心指导下,我们相继推出创刊号和神经系统罕见病、儿童罕见病、血液系统罕见病系列专刊,期刊网站和微信公众号也同步对外开放。创刊伊始,即受到热切关注,我刊已被中国学术期刊全文数据库、万方数据库等国内重要数据库收录,稿件下载、引用量和微信公众号阅读量迅速提升,学术影响力不断扩大。对于促进我国罕见病诊疗研究领域的学术交流,助力罕见病诊疗保障水平的提升发挥了积极作用。
展望来年,我们将密切追踪国内外罕见病研究领域的新动态和新成果,继续开展选题组稿工作,重点加强“述评、专家笔谈、指南与共识、多学科病例讨论、罕见病政策研究”等特色栏目建设,推动期刊学术质量迈上新的台阶。
心血管系统罕见病发病早、死亡率较高,患者长期面临诊断难和治疗率低的困境。提升心血管系统罕见病的诊疗水平,加速相关药物的研究和开发已日益受到关注。近年来,随着基础研究和新技术的迅猛发展,多种罕见心血管疾病的诊疗有了突破性进展。为此,本期特别以“心血管系统罕见病”为专题,系统阐述了家族性高胆固醇血症、轻链型淀粉样变性的心血管病、肥厚型心肌病、慢性血栓栓塞性肺动脉高压等多种罕见心血管疾病的诊断、治疗及新药研发进展,对《转甲状腺素蛋白心脏淀粉样变诊断与治疗中国专家共识》进行了解读及诊断路径更新,探讨分析了原发性心脏血管肉瘤等疾病的影像学特点,并分享了纯合子家族性高胆固醇血症患儿肝移植术治疗随访的多学科合作诊治经验。同时,今年我刊还将陆续发布青少年成人脊髓性肌萎缩症临床诊疗指南等临床实践指导性文章。籍以一系列重点专题的出版,为各学科领域的临床医师及相关专业研究人员提供参考和借鉴。
一元复始,万象更新。承载着新的希望和梦想,我们将与编委、专家、作者和读者们携手共进,立足前沿,探索创新,砥砺前行。为造福罕见病患者,推进“健康中国”战略实施发挥更大力量!

2023年1月
作者贡献:封思琴、唐牧云共同参与文献检索、分析与文章撰写;张抒扬和吴炜负责选题、修改及润色。利益冲突:所有作者均声明不存在利益冲突。 -
![]()
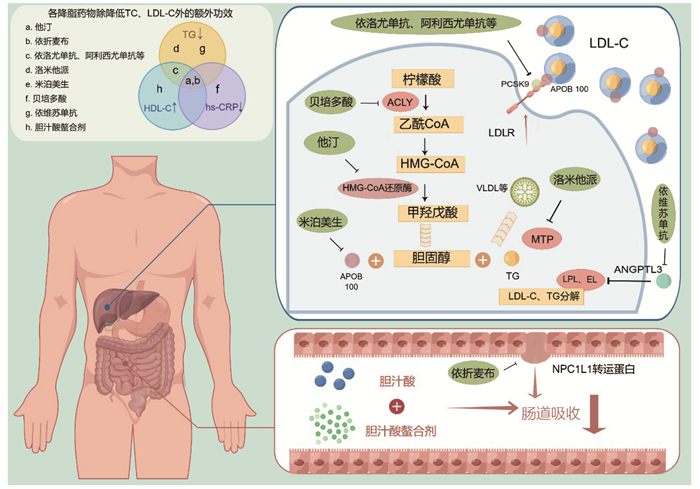
图 1 各降脂药物的作用机制
此图由Figdraw绘制;hs-CRP:超敏C反应蛋白;HMG-CoA还原酶:羟甲基戊二酰辅酶A还原酶;APOB 100:载脂蛋白B100;LPL:脂蛋白脂肪酶;EL:内皮脂肪酶;NPC1L1:类尼曼-匹克C1蛋白1
Figure 1. Mechanism of lipid-lowering drugs
表 1 与低密度脂蛋白胆固醇(LDL-C)水平相关基因
Table 1 Genes associated with low-density lipoprotein cholesterol(LDL-C)levels
基因 中文全称 染色体定位 突变数量* 功能简介 药物 常染色体(共)显性遗传 LDLR 低密度脂蛋白受体 19p13.3 2597 LDLR突变是FH发病的主要致病原因,该基因编码的LDLR细胞膜蛋白通过调控LDL的胞吞作用,可特异性识别LDL颗粒上的B100脂蛋白、乳糜微粒残体及中间密度脂蛋白上的APOE。其突变导致LDL无法进入细胞分解代谢,导致循环LDL-C水平增高,从而诱发动脉粥样硬化等一系列脂代谢紊乱。 现代降脂药物的基石靶点,他汀类药物、PCSK9抑制剂等最终均作用于此 APOB 载脂蛋白B 2p24~p23 492 APOB是乳糜微粒和VLDL中的关键结构蛋白,为食源性和内源性产生的胆固醇和甘油三酯的脂蛋白的组装和分布提供了基础结构。人类APOB基因编码单个RNA转录本,通过mRNA编辑从中翻译出2种亚型APOB-48和APOB-100。其中,APOB-48主要由肠细胞产生,是乳糜微粒的基本结构成分;而APOB-100主要由肝细胞产生,是VLDL和LDL的基本结构成分。APOB-100对LDL-LDLR复合物形成非常重要,APOB基因的功能缺失型突变会降低LDL与LDLR的亲和力从而促使FH的发生;此外,已被证实其突变可以导致低β脂蛋白血症[5]。 米泊美生 PCSK9 前蛋白转化酶枯草溶菌素9 1p32.3 144 PCSK9是继LDLR和APOB之后第3个与FH相关的基因[6]。PCSK9参与LDLR的负反馈调节,通过结合LDLR使其被转运至溶酶体进行降解,抑制LDLR再循环到细胞表面,导致LDL依赖性LDL-C水平的上调。其功能增强突变导致LDL-C水平升高,进而导致FH。 PCSK9抑制剂如依洛尤单抗、阿利西尤单抗和英克西兰 APOE 载脂蛋白E 19q13.2 86 APOE是LDLR家族的配体之一,其与血浆胆固醇水平变化密切相关[7]。 目前针对APOE的药物研发最高处于Ⅱ期临床阶段 HMGCR 3-羟基-3-甲基戊
二酰-CoA还原酶5q13.3~q14 6 HMGCR作为甾醇调节元件结合蛋白的调控基因,对细胞的胆固醇稳态具有至关重要的调节作用[8]。 他汀类 ACLY ATP-柠檬酸裂解酶 17q21.2 8 ATP-柠檬酸裂解酶是催化乙酰辅酶A产生的主要酶,乙酰辅酶A参与脂肪合成与胆固醇合成,发挥糖代谢与产生脂肪酸之间的桥梁作用[9]。 贝培多酸 ANGPTL3 血管生成素样
蛋白31p31.1~p22.3 58 血管生成素样蛋白3是脂蛋白脂肪酶和内皮脂肪酶的内源性抑制剂,已被证明可调节肝脏产生的VLDL水平,在人类脂蛋白代谢中起关键作用。ANGPTL3的致病变异已经在家族性混合型低胆固醇血症病例中被报道,这种疾病的特点是循环中VLDL、LDL和HDL的浓度极低[10]。 依维苏单抗 LIPC 肝脂酶 15q21~q23 51 LIPC功能获得性突变可改变肝脂肪酶的底物特异性,导致磷脂酶活性升高[11]。LIPC是继ANGPTL3之后第2个被发现的导致家族性混合型低胆固醇血症的基因[12]。 尚无 MTTP 微粒体甘油三酯
转运蛋白4q24 115 MTTP位于肠细胞和肝细胞的内质网中,在VLDL和乳糜微粒的产生中发挥重要作用。MTTP基因的突变会导致这些含APOB蛋白的脂蛋白组装障碍,从而导致无β脂蛋白血症,并影响脂肪和脂溶性维生素吸收[13]。 洛米他派 LIPG 内皮脂肪酶 18q21.1 47 内皮脂肪酶由血管内皮合成,参与HDL代谢[14]。 尚无 ABCA1 ATP结合盒A亚
家族成员19q31.1 331 Cenarro等[15]发现ABCA1的R219K多态性影响FH早发冠心病的风险。ABCA1是一种膜转运蛋白,可刺激胆固醇和磷脂外流至载脂蛋白A-I,在调节血浆HDL-C和载脂蛋白A-I代谢中起关键作用。 尚无 AKT1 AKT丝氨酸/苏氨酸激酶1 14q32.32 16 血管平滑肌细胞中的AKT1缺乏可诱使斑块纤维帽变薄,导致广泛的坏死核心区域形成[16]。 尚无 AMPD1 腺苷单磷酸脱氨酶1 1p13 17 AMPD1是嘌呤核苷代谢过程中起关键作用的酶。 尚无 APOA4 载脂蛋白A4 11q23 24 APOA4-360-2等位基因与FH患者对饮食反应的影响有关[17],APOA4是一种在小肠中合成的糖蛋白,并在脂肪吸收过程中与肠道脂蛋白一起分泌,其在富含甘油三酯的脂蛋白和HDL的代谢中起重要作用。 尚无 APOA5 载脂蛋白A5 11q23 97 APOA5基因多态性与中国高脂血症患者的基线甘油三酯水平相关[18]。载脂蛋白A5在调节血清甘油三酯浓度中起关键作用。 尚无 CBLC CBL原癌基因C 19q13.2 4 CBLC编码E3泛素连接酶,与细胞增殖有关。 尚无 C5AR2 补体成分5a受体2 19q13.33 4 补体成分5a受体是补体系统的一个重要组成成分。 尚无 GPIHBP1 糖基化磷脂酰肌醇锚定高密度脂蛋白结合蛋白1 8q24.3 51 GPIHBP1基因缺陷会影响乳糜微粒的分解代谢和肝脏对其残留物的摄取,从而导致高TG水平[19]。 尚无 常染色体隐性遗传 LDLRAP1 低密度脂蛋白受体衔接蛋白1 1p36~p35 54 LDLRAP1通过连接LDLR的细胞内域和网格蛋白,参与细胞LDL的吸收。 尚无 ABCG5 ATP结合盒G亚
家族成员52p21 115 ABCG5和ABCG8基因分别编码ATP结合盒转运蛋白G5和G8,其突变影响甾醇的跨膜转运,引起一种常染色体隐性遗传病——谷甾醇血症。这一类患者血浆植物甾醇和胆固醇水平严重升高,临床表型与FH相似。Reeskamp等[20]报道2.4%的FH患者携带致病性ABCG5和ABCG8基因突变,且较LDLR突变杂合子携带者LDL-C水平更低。 尚无 ABCG8 ATP结合盒G亚
家族成员82p21 119 功能同ABCG5 尚无 LIPA 溶酶体酸脂肪酶A 10q23.2~q23.3 123 LIPA突变可能引起罕见的隐性溶酶体贮积病——溶酶体酸性脂肪酶(LAL) 缺乏症,LAL功能障碍导致胆固醇酯和TG在肝细胞、肠和肾上腺等中积累。在FH群体中LIPA突变携带者的概率可能相对增高[21]。 尚无 *截至2022年9月27日,人类基因突变数据库结果(http://www.hgmd.cf.ac.uk/ac/index.php);FH:家族性高胆固醇血症;VLDL:极低密度脂蛋白;HDL-C: 高密度脂蛋白胆固醇;LAL:溶酶体酸性脂肪酶  下载: 导出CSV
下载: 导出CSV
表 2 初级卫生保健中按年龄筛查家族性高胆固醇血症(FH)的初步建议
Table 2 Tentative recommendations for screening by age for familial hypercholesterolemia(FH) in primary care
年龄(岁) 开展血清LDL-C水平检测的情况 开展基因检测的情况* 确诊后级联筛查范围 0~2 无需,除非父母双方均LDL-C≥3.6 mmol/L 父母双方均基因诊断阳性 父母和兄弟姐妹 3~11 >2岁且有家族史;无殊则在5 ~ 11岁之间进行 LDL-C≥3.6 mmol/L及阳性家族史 父母和兄弟姐妹 12~29 如果之前未进行过测试,最好在21岁之前进行 (1)LDL-C≥4.7 mmol/L
(2)皮肤/腱黄色瘤或脂性角膜弓(<45岁)
(3)一级亲属中有FH或早发ASCVD患者
满足以上3条中的1条者父母和兄弟姐妹 30~60 根据《中国成人血脂异常防治指南》[40],建议20~40岁成年人至少每5年测量1次血脂;40岁以上男性和绝经期后女性每年检测血脂 同上 所有一级亲属 >60 同上 同上 所有一级亲属 *需要排除继发性高胆固醇血症,其中家族史包括FH及早发ASCVD(男性<55岁或女性<65岁)的家族史
下载: 导出CSV
-
[1] Defesche JC, Gidding SS, Harada-Shiba M, et al. Familial hypercholesterolaemia[J]. Nat Rev Dis Primers, 2017, 3: 17093. doi: 10.1038/nrdp.2017.93
[2] Gidding SS, Champagne MA, de Ferranti SD, et al. The agenda for familial hypercholesterolemia: a scientific statement from the American Heart Association[J]. Circulation, 2015, 132(22): 2167-2192. doi: 10.1161/CIR.0000000000000297
[3] Khera AV, Won HH, Peloso GM, et al. Diagnostic yield and clinical utility of sequencing familial hypercholeste-rolemia genes in patients with severe hypercholesterolemia[J]. J Am Coll Cardiol, 2016, 67(22): 2578-2589. doi: 10.1016/j.jacc.2016.03.520
[4] Feng S, Zhao X, Wang Y, et al. Autosomal recessive hypercholesterolemia caused by a novel LDLRAP1 variant and membranous nephropathy in a Chinese girl: a case report[J]. Front Cardiovasc Med, 2022, 9: 811317. doi: 10.3389/fcvm.2022.811317
[5] Vega GL, Grundy SM. In vivo evidence for reduced binding of low density lipoproteins to receptors as a cause of primary moderate hypercholesterolemia[J]. J Clin Invest, 1986, 78(5): 1410-1414. doi: 10.1172/JCI112729
[6] Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia[J]. Nat Genet, 2003, 34(2): 154-156. doi: 10.1038/ng1161
[7] Marduel M, Ouguerram K, Serre V, et al. Description of a large family with autosomal dominant hypercholesterolemia associated with the APOE p. Leu167del mutation[J]. Hum Mutat, 2013, 34(1): 83-87. doi: 10.1002/humu.22215
[8] Soufi M, Ruppert V, Kurt B, et al. The impact of severe LDL receptor mutations on SREBP-pathway regulation in homozygous familial hypercholesterolemia (FH)[J]. Gene, 2012, 499(1): 218-222. doi: 10.1016/j.gene.2012.02.031
[9] Brandts J, Ray KK. Bempedoic acid, an inhibitor of ATP citrate lyase for the treatment of hypercholesterolemia: early indications and potential[J]. Expert Opin Investig Drugs, 2020, 29(8): 763-770. doi: 10.1080/13543784.2020.1778668
[10] Pirillo A, Catapano AL, Norata GD. Monoclonal antibodies in the management of familial hypercholesterolemia: focus on PCSK9 and ANGPTL3 inhibitors[J]. Curr Atheroscler Rep, 2021, 23(12): 79. doi: 10.1007/s11883-021-00972-x
[11] Dijk W, Di Filippo M, Kooijman S, et al. Identification of a gain-of-function LIPC variant as a novel cause of familial combined hypocholesterolemia[J]. Circulation, 2022, 146(10): 724-739. doi: 10.1161/CIRCULATIONAHA.121.057978
[12] Stankov S, Vitali C, Rader DJ. Gain-of-Function variants in lipid genes enhance biological insight and point toward therapeutic opportunities[J]. Circulation, 2022, 146(10): 740-742. doi: 10.1161/CIRCULATIONAHA.122.061233
[13] Takahashi M, Okazaki H, Ohashi K, et al. Current diagnosis and management of abetalipoproteinemia[J]. J Atheroscler Thromb, 2021, 28(10): 1009-1019. doi: 10.5551/jat.RV17056
[14] Corral P, Geller AS, Polisecki EY, et al. Unusual genetic variants associated with hypercholesterolemia in Argentina[J]. Atherosclerosis, 2018, 277: 256-261. doi: 10.1016/j.atherosclerosis.2018.06.009
[15] Cenarro A, Artieda M, Castillo S, et al. A common variant in the ABCA1 gene is associated with a lower risk for premature coronary heart disease in familial hypercholesterolaemia[J]. J Med Genet, 2003, 40(3): 163-168. doi: 10.1136/jmg.40.3.163
[16] Rotllan N, Wanschel AC, Fernández-Hernando A, et al. Genetic evidence supports a major role for Akt1 in VSMCs during atherogenesis[J]. Circ Res, 2015, 116(11): 1744-1752. doi: 10.1161/CIRCRESAHA.116.305895
[17] Carmena-Ramón R, Ascaso JF, Real JT, et al. Genetic variation at the apoA-IV gene locus and response to diet in familial hypercholesterolemia[J]. Arterioscler Thromb Vasc Biol, 1998, 18(8): 1266-1274. doi: 10.1161/01.ATV.18.8.1266
[18] Hu M, Mak VW, Tomlinson B. Polymorphisms in apolipoprotein E and apolipoprotein A-V do not influence the lipid response to rosuvastatin but are associated with baseline lipid levels in Chinese patients with hyperlipidemia[J]. J Clin Lipidol, 2012, 6(6): 585-592. doi: 10.1016/j.jacl.2012.02.005
[19] Kwiterovich PO Jr. Diagnosis and management of familial dyslipoproteinemias[J]. Curr Cardiol Rep, 2013, 15(6): 371. doi: 10.1007/s11886-013-0371-5
[20] Reeskamp LF, Volta A, Zuurbier L, et al. ABCG5 and ABCG8 genetic variants in familial hypercholesterolemia[J]. J Clin Lipidol, 2020, 14(2): 207-217.e7. doi: 10.1016/j.jacl.2020.01.007
[21] Sjouke B, Defesche JC, de Randamie JSE, et al. Sequencing for LIPA mutations in patients with a clinical diagnosis of familial hypercholesterolemia[J]. Atherosclerosis, 2016, 251: 263-265. doi: 10.1016/j.atherosclerosis.2016.07.008
[22] Cui Y, Li S, Zhang F, et al. Prevalence of familial hypercholesterolemia in patients with premature myocardial infarction[J]. Clin Cardiol, 2019, 42(3): 385-390. doi: 10.1002/clc.23154
[23] Chen P, Chen X, Zhang S. Current status of familial hypercholesterolemia in China: a need for patient FH registry systems[J]. Front Physiol, 2019, 10: 280. doi: 10.3389/fphys.2019.00280
[24] Zhang S, Chen L, Zhang Z, et al. Orphan drug development in China: progress and challenges[J]. Lancet, 2019, 394(10204): 1127-1128. doi: 10.1016/S0140-6736(19)32179-8
[25] Humphries SE, Whittall RA, Hubbart CS, et al. Genetic causes of familial hypercholesterolaemia in patients in the UK: relation to plasma lipid levels and coronary heart disease risk[J]. J Med Genet, 2006, 43(12): 943-949. doi: 10.1136/jmg.2006.038356
[26] Harada-Shiba M, Arai H, Ishigaki Y, et al. Guidelines for diagnosis and treatment of familial hypercholesterolemia 2017[J]. J Atheroscler Thromb, 2018, 25(8): 751-770. doi: 10.5551/jat.CR003
[27] Kramer AI, Akioyamen LE, Lee S, et al. Major adverse cardiovascular events in homozygous familial hypercholesterolaemia: a systematic review and meta-analysis[J]. Eur J Prev Cardiol, 2022, 29(5): 817-828. doi: 10.1093/eurjpc/zwab224
[28] Talmud PJ, Shah S, Whittall R, et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study[J]. Lancet, 2013, 381(9874): 1293-1301. doi: 10.1016/S0140-6736(12)62127-8
[29] Sturm AC, Knowles JW, Gidding SS, et al. Clinical genetic testing for familial hypercholesterolemia: JACC scientific expert panel[J]. J Am Coll Cardiol, 2018, 72(6): 662-680. doi: 10.1016/j.jacc.2018.05.044
[30] Galema-Boers JM, Versmissen J, Roeters van Lennep HW, et al. Cascade screening of familial hypercholesterolemia must go on[J]. Atherosclerosis, 2015, 242(2): 415-417. doi: 10.1016/j.atherosclerosis.2015.07.020
[31] Varret M, Abifadel M, Rabès JP, et al. Genetic heterogeneity of autosomal dominant hypercholesterolemia[J]. Clin Genet, 2008, 73(1): 1-13. doi: 10.1111/j.1399-0004.2007.00915.x/pdf
[32] Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group[J]. BMJ, 1991, 303(6807): 893-896. doi: 10.1136/bmj.303.6807.893
[33] Umans-Eckenhausen MA, Defesche JC, Sijbrands EJ, et al. Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands[J]. Lancet, 2001, 357(9251): 165-168. doi: 10.1016/S0140-6736(00)03587-X
[34] 中华医学会心血管病学分会动脉粥样硬化及冠心病学组, 中华心血管病杂志编辑委员会. 家族性高胆固醇血症筛查与诊治中国专家共识[J]. 中华心血管病杂志, 2018, 46(2): 99-103. doi: 10.3760/cma.j.issn.0253-3758.2018.02.006 [35] Andermann A, Blancquaert I, Beauchamp S, et al. Revisiting Wilson and Jungner in the genomic age: a review of screening criteria over the past 40 years[J]. Bull World Health Organ, 2008, 86(4): 317-319. doi: 10.2471/BLT.07.050112
[36] Brett T, Qureshi N, Gidding S, et al. Screening for familial hypercholesterolaemia in primary care: time for general practice to play its part[J]. Atherosclerosis, 2018, 277: 399-406. doi: 10.1016/j.atherosclerosis.2018.08.019
[37] Representatives of the Global Familial Hypercholesterolemia C, Wilemon KA, Patel J, et al. Reducing the clinical and public health burden of familial hypercholesterolemia: a global call to action[J]. JAMA Cardiol, 2020, 5(2): 217-229. doi: 10.1001/jamacardio.2019.5173
[38] deGoma EM, Ahmad ZS, O'Brien EC, et al. Treatment gaps in adults with heterozygous familial hypercholesterolemia in the United States: data from the CASCADE-FH registry[J]. Circ Cardiovasc Genet, 2016, 9(3): 240-249. doi: 10.1161/CIRCGENETICS.116.001381
[39] Jiang L, Stoekenbroek RM, Zhang F, et al. Homozygous familial hypercholesterolemia in China: genetic and clinical characteristics from a real-world, multi-center, cohort study[J]. J Clin Lipidol, 2022, 16(3): 306-314. doi: 10.1016/j.jacl.2022.03.003
[40] Joint committee issued Chinese guideline for the management of dyslipidemia in adults. 2016 Chinese guideline for the management of dyslipidemia in adults[J]. Zhonghua Xin Xue Guan Bing Za Zhi, 2016, 44(10): 833-853. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_lnxzbxzz-e201801001
[41] Antoniazzi L, Arroyo-Olivares R, Bittencourt MS, et al. Adherence to a Mediterranean diet, dyslipidemia and inflammation in familial hypercholesterolemia[J]. Nutr Metab Cardiovasc Dis, 2021, 31(7): 2014-2022. doi: 10.1016/j.numecd.2021.04.006
[42] Miedema MD, Nauffal VD, Singh A, et al. Statin therapy for young adults: a long-term investment worth considering[J]. Trends Cardiovasc Med, 2020, 30(1): 48-53. doi: 10.1016/j.tcm.2019.02.004
[43] Pang J, Chan DC, Watts GF. The knowns and unknowns of contemporary statin therapy for familial hypercholesterolemia[J]. Curr Atheroscler Rep, 2020, 22(11): 64. doi: 10.1007/s11883-020-00884-2
[44] Besseling J, Hovingh GK, Huijgen R, et al. Statins in familial hypercholesterolemia: consequences for coronary artery disease and all-cause mortality[J]. J Am Coll Cardiol, 2016, 68(3): 252-260. doi: 10.1016/j.jacc.2016.04.054
[45] Raal FJ, Pilcher GJ, Panz VR, et al. Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy[J]. Circulation, 2011, 124(20): 2202-2207. doi: 10.1161/CIRCULATIONAHA.111.042523
[46] Catapano AL, Graham I, De Backer G, et al. 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias[J]. Eur Heart J, 2016, 37(39): 2999-3058. doi: 10.1093/eurheartj/ehw272
[47] Park K, Vishnevetskaya K, Vaidyanathan J, et al. Pediatric drug development studies for familial hypercholesterolemia submitted to the US Food and Drug Administration between 2007 and 2020[J]. J Clin Pharmacol, 2022, 62(3): 397-408. doi: 10.1002/jcph.1973
[48] Writing Committee, Lloyd-Jones DM, Morris PB, et al. 2022 ACC expert consensus decision pathway on the role of nonstatin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the American College of Cardiology Solution Set Oversight Committee[J]. J Am Coll Cardiol, 2022, 80(14): 1366-1418. doi: 10.1016/j.jacc.2022.07.006
[49] Raal FJ, Hovingh GK, Catapano AL. Familial hypercholesterolemia treatments: guidelines and new therapies[J]. Atherosclerosis, 2018, 277: 483-492. doi: 10.1016/j.atherosclerosis.2018.06.859
[50] Ziółkowska S, Kijek N, Zendran I, et al. Familial hypercholesterolemia-treatment update in children, systematic review[J]. Pediatr Endocrinol Diabetes Metab, 2022, 28(2): 152-161. doi: 10.5114/pedm.2022.116112
[51] Hammersley D, Signy M. Ezetimibe: an update on its clinical usefulness in specific patient groups[J]. Ther Adv Chronic Dis, 2017, 8(1): 4-11. doi: 10.1177/2040622316672544
[52] Benekos T, Kosmeri C, Vlahos A, et al. Nine-year overview of dyslipidemia management in children with heterozygous familial hypercholesterolemia: a university hospital outpatient lipid clinic project in Northwestern Greece[J]. J Pediatr Endocrinol Metab, 2020, 33(4): 533-538. doi: 10.1515/jpem-2019-0250
[53] Iannuzzo G, Buonaiuto A, Calcaterra I, et al. Association between causative mutations and response to PCSK9 inhibitor therapy in subjects with familial hypercholesterolemia: a single center real-world study[J]. Nutr Metab Cardiovasc Dis, 2022, 32(3): 684-691. doi: 10.1016/j.numecd.2021.10.025
[54] Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for homozygous familial hypercholesterolemia[J]. N Engl J Med, 2020, 383(8): 711-720. doi: 10.1056/NEJMoa2004215
[55] 陈本川. 降胆固醇新药——贝培多酸(bempedoic acid)及其与依折麦布(ezetimibe)的复方片剂[J]. 医药导报, 2020, 39(10): 1448-1457. https://www.cnki.com.cn/Article/CJFDTOTAL-YYDB202010032.htm [56] Alonso R, Cuevas A, Mata P. Lomitapide: a review of its clinical use, efficacy, and tolerability[J]. Core Evid, 2019, 14: 19-30. doi: 10.2147/CE.S174169
[57] D'Erasmo L, Steward K, Cefalù AB, et al. Efficacy and safety of lomitapide in homozygous familial hypercholesterolaemia: the pan-European retrospective observational study[J]. Eur J Prev Cardiol, 2022, 29(5): 832-841. doi: 10.1093/eurjpc/zwab229
[58] Chambergo-Michilot D, Alur A, Kulkarni S, et al. Mipomersen in familial hypercholesterolemia: an update on health-related quality of life and patient-reported outcomes[J]. Vasc Health Risk Manag, 2022, 18: 73-80. doi: 10.2147/VHRM.S191965
[59] Astaneh B, Makhdami N, Astaneh V, et al. The effect of mipomersen in the management of patients with familial hypercholesterolemia: a systematic review and meta-analysis of clinical trials[J]. J Cardiovasc Dev Dis, 2021, 8(7): 82. doi: 10.3390/jcdd8070082
[60] Pederiva C, Capra ME, Viggiano C, et al. Early preven-tion of atherosclerosis: detection and management of hypercholesterolaemia in children and adolescents[J]. Life (Basel), 2021, 11(4): 345.
[61] Luirink IK, Hutten BA, Greber-Platzer S, et al. Practice of lipoprotein apheresis and short-term efficacy in children with homozygous familial hypercholesterolemia: data from an international registry[J]. Atherosclerosis, 2020, 299: 24-31. doi: 10.1016/j.atherosclerosis.2020.01.031
[62] Kayikcioglu M, Kuman-Tunçel O, Pirildar S, et al. Clinical management, psychosocial characteristics, and quality of life in patients with homozygous familial hypercholesterolemia undergoing LDL-apheresis in Turkey: results of a nationwide survey(A-HIT1 registry)[J]. J Clin Lipidol, 2019, 13(3): 455-467. doi: 10.1016/j.jacl.2019.02.001
[63] Al Dubayee M, Kayikcioglu M, van Lennep JR, et al. Is liver transplant curative in homozygous familial hypercholesterolemia? a review of nine global cases[J]. Adv Ther, 2022, 39(6): 3042-3057. doi: 10.1007/s12325-022-02131-3
[64] Mlinaric M, Bratanic N, Dragos V, et al. Case report: liver transplantation in homozygous familial hypercholesterolemia (HoFH)-long-term follow-up of a patient and literature review[J]. Front Pediatr, 2020, 8: 567895. doi: 10.3389/fped.2020.567895
[65] Vuorio A, Kovanen PT, Santos RD, et al. Prevention of cardiovascular burden in COVID-19 patients suffering from familial hypercholesterolemia: a global challenge[J]. Cardiol Ther, 2022, 11(1): 1-7. doi: 10.1007/s40119-021-00245-3
-
期刊类型引用(1)
1. 焦建,董薇,常智,张颖,李全,李珺奇,米宏志. 门控心肌灌注显像预测纯合子家族性高胆固醇血症患者主要心脏不良事件的研究. 心肺血管病杂志. 2024(06): 623-628 .  百度学术
百度学术
其他类型引用(0)
计量
- 文章访问数: 661
- HTML全文浏览量: 288
- PDF下载量: 159
- 被引次数: 1


