 作者投稿
作者投稿 专家审稿
专家审稿 编辑办公
编辑办公 主编办公
主编办公
Practice of the Qualification and Recognition for Orphan Drugs in the World and its Inspiration
-
摘要:
分析目前国内外罕见病药品资格认定现状,包括其负责机构、认定流程、认定及上市数量等。通过横向对比,分析典型发达国家和部分发展中国家在罕见病药品资格认定方面的差异,探究中国在罕见病药品监管方面的不足。借鉴典型发达国家和部分发展中国家的先进经验,以期为中国罕见病药品认定管理提供建议,为加快中国罕见病药品研发上市,为最大程度提高患者治疗可及性提供参考。
Abstract:We have analyzed the current status of recognization and qualification of orphan drugs in China and abroad, looking at the aspects of the authority institutions, identification and qualification process, and the number of orphan drugs identified and available in the market. By comparing and analyzing horizontally the differences in orphan drugs identification between representative developed countries vs. some developing countries, we discuss the inadequacy of orphan drugs supervision in China. We introduce the advanced experience from the developed countries and some developing countries to provide suggestions for the identification and management of orphan drugs, hoping to speed up the process of development and market availability of orphan drugs and to maximize patient's accessibility to treatment in China.
-
听力损失是常见的出生缺陷之一,发病率为1‰~3‰,致病因素中遗传因素约占60%。根据是否伴有其他症状,遗传性耳聋可分为非综合征耳聋和综合征耳聋,其中综合征耳聋属于罕见病,约占耳聋的30%,且存在遗传异质性,既有常染色体显性遗传和常染色体隐性遗传,也有伴性遗传和线粒体母系遗传,还具有不完全外显和外显不全的特点,临床表型不定,家族内成员和家系之间呈现的表型差异导致临床诊断难度增加,非常容易误诊和漏诊[1-2]。
目前已经发现超过400个综合征伴有耳聋的表现[3],相对比较常见的11个综合征耳聋的致病基因有48种[4]。这些基因编码的蛋白质包括离子通道蛋白、膜蛋白、转录因子和结构蛋白等。一部分相对常见的致病基因已经包含在耳聋Panel的临床检测中,但是还有很多综合征病因复杂、无明确的致病基因。罕见综合征耳聋的研究多为临床散发病例,伴耳聋的综合征罕见病例的分子遗传学还未见详细和系统的报道。本研究利用全外显子家系组(“trio”)测序方法分析致聋基因并探讨其在罕见综合征聋病诊疗中的应用,为罕见聋病综合征患者提供病因学分析及遗传学诊断与咨询。
1. 资料与方法
1.1 一般资料
对2021年1—12月间在首都医科大学附属北京儿童医院就诊的34例耳聋患儿的临床资料进行回顾性分析。所有研究对象均经过详细病史询问和一般体格检查,并完成系统听力学评估、内耳CT平扫和冠状位薄层CT检查,排除单侧耳聋及中耳炎、脑膜炎、占位性病变、外伤导致的听力损失。经上述综合评估,纳入研究对象均诊断为双耳感音神经性听力损失。本研究经首都医科大学附属北京儿童医院医学伦理委员会审核通过(审批号:[2021]-E-013-Y),临床资料收集及标本采集均获得患儿家长的知情同意,并签署知情同意书。
1.2 听力学评估
听力学评估遵循测试组合、交叉验证的原则进行。测试涵盖听觉生理和行为听力测试,具体包括:听性脑干诱发反应(auditory brainstem response, ABR)、稳态听觉诱发反应(auditory steady state response, ASSR)、畸变产物耳声发射(distort production of acoustic emission, DPOAE)、声导抗和行为测听。听力评估结束后对结果进行交叉验证,受试儿童各测试结果间具有良好的一致性。听力损失程度的判断基于以下原则:2岁以下儿童以气导ABR的波V反应阈判定听力损失程度,≤30 dB nHL为正常,31~50 dB nHL为轻度听力损失,51~70 dB nHL为中度听力损失,71~90 dB nHL为重度听力损失,>90 dB nHL为极重度听力损失。2岁及以上儿童以行为听力测试4个频率(500 Hz、1000 Hz、2000 Hz、4000 Hz)听阈平均值判定听力损失程度,25~40 dB HL为轻度听力损失,41~60 dB HL为中度听力损失,61~80 dB HL为重度听力损失,>80 dB HL为极重度听力损失。
1.3 基因检测方法及流程
提取患儿及其父母外周静脉血的基因组DNA,利用Illumina NovaSeq6000测序平台完成全外显子高通量测序,对人类基因组中约2万个基因的外显子区及临近剪切区的DNA利用安捷伦V6芯片进行捕获和富集,测序模式为150 PE,每个样品产生不低于10~12 Gb数据,数据质量平均Q30>85%,数据平均覆盖100 X。本次检测基于患儿及其父母的临床表型,利用Human Gene Mutation Database(HGMD)、Online Mendelian Inheritance in Man(OMIM)以及Gene4HL等数据库中收录的相关致病基因与变异,根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)遗传变异分类标准与指南,对比中国人群基因变异数据库进行生物信息学分析与医学解读,筛选致病的(pathogenic)、可能致病的(likely pathogenic)和临床意义未明的(uncertain)变异。同时对数据库中无记载的突变使用SIFT(Sorting Intolerant From Tolerant,http://sift.jcvi.org)、Polyphen2(http://genetics.bwh.harvard.edu/pph2/)等软件对变异致病性进行预测分析。
2. 结果
借助全外显子组测序对34例诊断为感音神经性听力损失患儿进行基因检测,其中男20例,女14例,年龄4月龄至11岁,受试者基本情况和检出情况见表 1。确诊了15例为常染色体隐性遗传的非综合征耳聋,包括GJB2基因突变8例,SLC26A4基因突变5例,MYO15A基因突变2例,检出的突变位点见表 2。令人意外的发现4例患儿为综合征耳聋,包括HARS2基因突变导致的Perrault综合征2型1例、USH2A基因突变导致的Usher综合征ⅡA型1例、GATA3基因突变导致的甲状旁腺功能减退-感音神经性耳聋-肾发育不良(hypoparathyroidism, sensorineural deafness, renal dysplasia, HDR)综合征1例、MITF基因突变导致的Waardenburg综合征Ⅰ型1例(表 3)。HARS2和USH2A基因突变导致的综合征为常染色体隐性遗传,患儿父母分别为突变携带者。GATA3和MITF基因突变导致的综合征为常染色体显性遗传,家系分析证实GATA3基因的c.1327delA突变为新发突变,患儿父母均正常;MITF基因的c.627C>A突变来自患儿父亲,但其父没有耳聋等临床症状。本组患者耳聋的分子病因确诊率达到56%(19/34),其中综合征耳聋占21%(4/19),其余15例经家系分析未发现明确的致病基因。
表 1 34例儿童基本情况及听力损失分级Table 1. Basic information and degree of hearing loss of 34 children指标 检出致病变异例数
[n(%)]未检出致病变异人数
[n(%)]年龄(岁) <1 5(14.71) 3(8.82) 1~3 9(26.47) 4(11.76) 4~6 3(8.82) 6(17.65) >6 2(5.88) 2(5.88) 性别 男 12(35.29) 7(20.59) 女 7(20.59) 8(23.53) 听力损失程度 轻度 1(2.94) 0 中度 6(17.65) 3(8.82) 重度 5(14.71) 4(11.76) 极重度 5(14.71) 5(14.71) 双耳不对称 2(5.88) 3(8.82) 影像学检查 内耳及听神经异常 8(23.53) 8(23.53) 未见异常 11(32.35) 7(20.59) 表 2 全外显子组测序确诊的非综合征耳聋致病基因和例数Table 2. The genes and cases of non-syndromic hearing loss diagnosed by whole-exome sequencing致病基因(转录本) 突变位点 检出例数 GJB2(NM_004004) c.235delC/c.235delC 2 c.235delC/c.35dupG 2 c.235delC/ c.299_300delAT 1 c.299_300delAT/c.257C>G 1 c.299_300delAT/ c.427C>T 1 c.235delC/c.109G>A 1 SLC26A4(NM_000441) IVS7-2A>G/ IVS7-2A>G 4 IVS7-2A>G/ c.2168A>G 1 MYO15A(NM_016239) c.855dupT/c.8033_8057delinsG 1 c.3505C>T/c.8158G>A 1 合计 15 表 3 全外显子组测序确诊的综合征耳聋遗传学和临床特征(n=4)Table 3. The genetic and clinical features of syndromic hearing loss diagnosed by whole-exome sequencing(n=4)编号 性别 年龄 致病基因
(转录本)遗传方式 突变位点 ACMG变异致病性分级 综合征 听力损失程度 干预方式 累及系统 13 男 3岁 HARS2(NM_012208) AR c.435_437del/
c.1403G>CLP/Unc Perrault综合征2型 中度 助听器 中枢和外周神经 15 女 11岁 USH2A(NM_206933) AR c.8559-2A>G/
c.11389+1delP/P Usher综合征ⅡA型 中度 助听器 眼(视网膜) 16 女 8岁 GATA3(NM_001002295) AD c.1327delA/-
(新发突变)LP 甲状旁腺功能减退-感音神经性耳聋-肾发育不良(HDR)综合征 中度 助听器 甲状旁腺/肾脏/骨骼/牙齿 22 女 8个月 MITF(NM_198159) AD c.627C>A/-
(来自父亲)LP Waardenburg综合征Ⅰ型 极重度 人工耳蜗 皮肤/虹膜/头发色素 AR: 常染色体隐性遗传; AD: 常染色体显性遗传; HDR: 甲状旁腺功能减退—感音神经性耳聋—肾发育不良; P: 致病的; LP: 可能致病的; Unc: 临床意义未明的 3. 讨论
在实施人类基因组计划之前,罕见病的诊断是极其困难的。最近10年中,全外显子组和基因组测序技术逐渐成熟并在临床广泛应用,显著提高了诊断率,拓宽了与遗传变异相关的疾病谱,为罕见病的诊断提供了新的选择和希望,从而为潜在的治疗方法提供了可能性。罕见病的罕有或非常少见的特点造成了临床病例的稀缺性,相关文献资料多数是病案报道,不同专业人员的观察重点不同导致报道的数据偏差,影响报道的全面性和客观性。对于罕见病的临床判断是非常困难的,特别是在基层,经常出现“当面不识君”的情况。
本组34例感音神经性听力损失患儿,基因检测前均未发现或报告其他异常体征。借助全外显子组测序确诊了19例中的4例由综合征耳聋相关基因突变导致,这个发现具有非常重要的临床意义:①明确了临床诊断、分型和分子病因;②为遗传咨询和婚育指导提供了依据;③提示进一步相关器官和功能检查的必要性;④为疾病的自然发展和干预提供了信息;⑤丰富了罕见基因病的表型谱。这4例患者分别为HARS2基因突变导致的Perrault综合征2型1例、USH2A基因突变导致的Usher综合征Ⅱ A型1例、GATA3基因突变导致的HDR综合征1例、MITF基因突变导致的Waardenburg综合征Ⅰ型1例。前2例为常染色体隐性遗传;后2例为常染色体显性遗传,其中1例为新发突变,另外1例的突变虽然来自父亲,但其父亲没有耳聋,所以这4例均表现为散发病例。
由于外显不全、临床症状不明显、性别和年龄等原因,儿童的综合征耳聋很容易误判为非综合征耳聋。本研究中13号病例是HARS2基因突变导致的Perrault综合征2型,患儿为男性,其哥哥也是耳聋,该综合征主要临床表现为耳聋及女性卵巢发育不全[5-7],目前关于男性的表型报道很少。USH2A基因突变导致的Usher综合征ⅡA型主要表现为耳聋和视网膜色素变性[8],视网膜色素变性多发生在10多岁,主要表现为渐进性的视野缺失和视力障碍。本研究中15号病例是USH2A基因突变导致的Usher综合征ⅡA型,年龄11岁,虽然目前没有相关视野的异常,但基因的结果提醒家长完善眼部的检查,有助于提早发现和干预。GATA3基因突变导致的HDR综合征,除了耳聋之外,还有甲状旁腺激素分泌不足导致的钙代谢异常和肾脏的发育或功能异常[9]。本研究中16号病例是GATA3基因突变导致的HDR综合征,年龄8岁,表型除了额头大之外,无明显的钙代谢异常,也未做过肾脏的B超和功能检查。基因的结果提示需要进一步检查甲状旁腺激素水平和血钙浓度、腹部B超和肾功能。Waardenburg综合征Ⅰ型主要表现为耳聋和色素异常,包括虹膜、头发和皮肤的颜色异常。本研究中22号病例是MITF基因突变导致的Waardenburg综合征Ⅰ型,但是除了面色白皙,无其他明显色素异常,其父亲也携带该基因突变,回顾性问诊和查体证实其父亲有少白头和皮肤白皙,但最初问诊时因这两个表征在正常人群中比较常见,没有考虑到可能相关的综合征。由此可见,仅依靠临床表现和特征诊断综合征耳聋非常困难,极易漏诊,基因诊断对明确耳聋的病因不可或缺,检测结果是患儿及其家庭进行遗传咨询的重要依据,特别是对婚育和再生育的指导,并为产前诊断或第三代试管婴儿胚胎移植前遗传学诊断(preimplantation genetic diagnosis,PGD)技术提供了目标基因。
耳聋最常见致病基因是GJB2和SLC26A4,这两个基因已经包含在新生儿耳聋基因筛查范围内,也是耳聋基因检测的候选目标基因,本组34例患者中有13例是这两个基因突变导致,占38%(13/34)。因此,在耳聋的基因检测方案中,可以先做这两个基因的检测,然后再进行Panel或全外显子基因检测寻找少见或罕见的致病基因,这样分步骤的检测流程与本文的直接全外显子组测序相比,其性价比会更高一些。MYO15A基因突变在本组病例中发现了2例,是导致遗传性耳聋的第3个常见耳聋基因[10],但目前尚未纳入新生儿耳聋基因筛查项目中[11]。Fu等[12]对中国81例MYO15A基因致聋患者的基因型和表型进行了分析,发现双等位基因非截短突变所占比例较少(12/81),但多数(10/12)表现为极重度耳聋。本文中的2例MYO15A基因突变致聋患儿都是携带一个截短突变和一个非截短突变,其中一例患儿携带MYO15A基因c.855dupT(p.P286Sfs*15)/c.8033_8057delinsG (p.N2678_ D2686delinsS) 复合杂合突变,表现为双耳中度耳聋;另外一例患儿携带MYO15A基因c.3505C>T(p.R1169X)/c.8158G>A(p.D2720N),表现为左耳重度耳聋,右耳极重度耳聋。本研究发现的MYO15A基因c.8033_ 8057delinsG(p.N2678_ D2686delinsS)突变为首次报道,丰富了该基因的突变谱。
全外显子基因检测的最佳实践方案是家系检测(“trio”),即包括先证者及其父母,比较3人的测序结果有助于分析罕见变异的背景和来源。本组中有15例虽然基因测序结果提示了可能致病基因,但经家系分析否定了其致病相关性,如果没有父母的基因结果,测序结果的分析更加困难,不确定性也会明显增加。全外显子基因检测对于确诊罕见的综合征和非综合征耳聋都非常有意义,检测效率高,该检测的优势还包括应用范围广,数据可以长期保留,在今后需要时再进行分析,但也有一定的局限性,如只能检测有限的拷贝数变异,不能检测内含子变异(除了目标外显子的侧翼),也不能检测三核苷酸重复扩增、甲基化异常。因此,对于检测结果阴性的病例,结合临床表现,还可以选择相应的检测,进一步完善遗传学分析和诊断。
作者贡献:朱晓红负责检索文献,撰写、修订论文;李顺平提出选题思路,并负责修订、审校论文;陈敬丹、冯俊超、张海琴、刘嘉琪、谢诗瑶、张悦提出修订意见,辅助撰写论文。利益冲突:所有作者均声明不存在利益冲突。 -
![]()
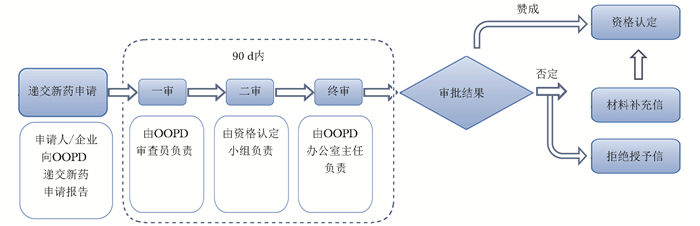
图 1 美国罕见病药品认定流程
OOPD:罕见病药品研发办公室
Figure 1. Identification process of rare diseases drugs in the United States
![]()
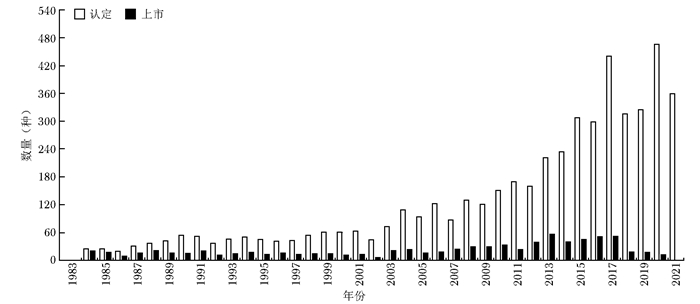
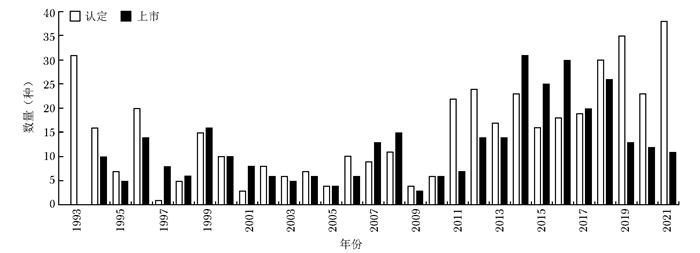
图 2 美国罕见病药品认定与上市数量
Figure 2. Identification and marketing quantity of rare disease drugs in the United States
![]()
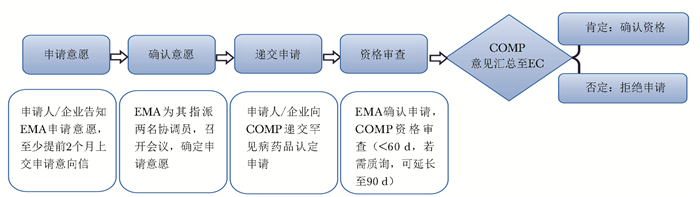
图 3 欧盟罕见病药品认定流程
EMA:欧洲药品管理局;COMP:罕见病药品委员会;EC:欧盟委员会
Figure 3. Identification process of rare diseases drugs in EU
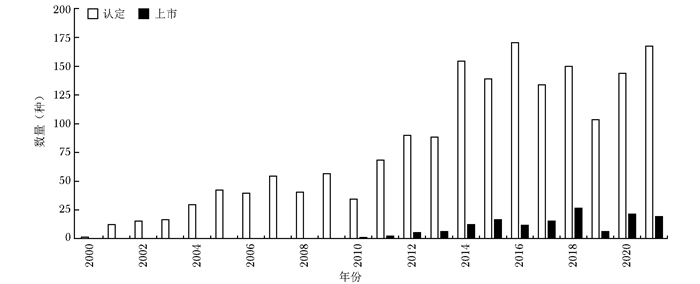
![]()
图 4 欧盟罕见病药品认定与上市数量
Figure 4. Identification and marketing quantity of rare disease drugs in EU
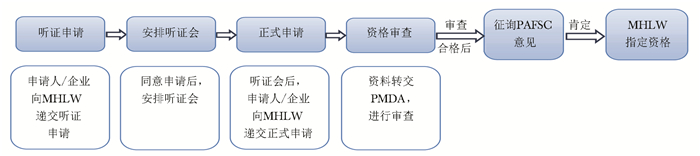
![]()
图 5 日本罕见病药品认定流程
MHLW:日本厚生劳动省;PMDA:医药品医疗器械综合管理机构;PAFSC:药事和食品卫生委员会
Figure 5. Identification process of rare disease drugs in Japan
![]()
图 6 日本罕见病药品认定与上市数量
Figure 6. Identification and marketing quantity of rare disease drugs in Japan
表 2 部分发展中国家罕见病领域发展汇总
Table 2 Summary of development for rare diseases in some developing countries
国家 罕见病定义 罕见病药品认定标准 专项立法 认定监管部门 研发激励政策 印度 无* 治疗<50万患者的药物 无* 无* 国家罕见病治疗政策;资金支持 墨西哥 采用欧盟定义 无* 《普通卫生法》224条修订 无* 加速批准 巴西 患病率低于0.65‰的疾病 无* 无* 无* 罕见病国家政策;优先审查 哥伦比亚 患病率低于0.2‰的疾病 无* 第1392(2010)号法律 无* 无* 秘鲁 无* 采用欧盟定义 第29698(2011)号法律 无* 无* 阿根廷 采用欧盟定义 无* 第26.689(2011)号法律 无* 无* *截至2021年12月31日尚未颁布相关文件  下载: 导出CSV
下载: 导出CSV
表 1 典型发达国家及地区罕见病药品认定情况汇总
Table 1 Summary of drug identification for rare diseases in typical developed countries and regions
分类 美国 欧盟 日本 罕见病定义 患病人数低于20万(总人口的0.75‰)或超过20万无商业回报的疾病 低于0.5‰;严重性、危及生命的、退化性;无可替代治疗的疾病 患病人数低于5万人(总人口0.4‰);病因不明;无有效治疗方式;严重经济、心理负担的疾病 立法基础 《孤儿药法案》(Orphan Drug Act,ODA) 《罕见病药品条例》(Regulation(EC) No 141/2000 on orphan medicinal products,EC 141/2000) 《药事法》(Pharmaceutical Affairs Law) 监管部门 FDA EMA MHLW 认定标准 用于预防、诊断、治疗美国罕见病的药物 用于预防、诊断、治疗欧盟罕见病的药物 治疗日本罕见病;具备清楚的药品研发计划和支持上市的科学依据 认定机构 OOPD COMP MHLW 认定总数(个)* 5049 1762 438 上市总数(个)* 859 151 344 上市数量占比(%) 17.0 8.6 78.5 *认定总数与上市总数的时间范围:本国法案颁布之日至2021年12月31日;上市数量占比:上市总数/认定总数×100%;EC:欧盟委员会,FDA:美国食品药品监督管理局,EMA:欧洲药品管理局,MHLW:日本厚生劳动省,OOPD:罕见病药品研发办公室,COMP:罕见病药品委员会
下载: 导出CSV
-
[1] Martínez-deMiguel C, Segura-Bedmar I, Chacón-Solano E, et al. The RareDis corpus: a corpus annotated with rare diseases, their signs and symptoms[J]. J Biomed Inform, 2022, 125: 103961. doi: 10.1016/j.jbi.2021.103961
[2] Chan AYL, Chan VKY, Olsson S, et al. Access and unmet needs of orphan drugs in 194 countries and 6 areas: A Global Policy Review With Content Analysis[J]. Value Health, 2020, 23: 1580-1591. doi: 10.1016/j.jval.2020.06.020
[3] Belousova Olga A, Groen Aard J, Ouendag Aniek M. Opportunities and barriers for innovation and entrepreneurship in orphan drug development[J]. Technological Forecasting & Social Change, 2020, 161: 120333.
[4] 冀希炜, 梁家彬, 季双敏. 罕见病治疗国内外的研究现状[J]. 中国临床药理学杂志, 2019, 3: 305-308. https://www.cnki.com.cn/Article/CJFDTOTAL-GLYZ201903032.htm [5] Food and Drug Administration. Orphan Drug Act-Relevant Excerpts[EB/OL]. (2018-09-03)[2022-05-08]. https://www.fda.gov/industry/designating-orphan-product-drugs-and-biological-products/orphan-drug-act-relevant-excerpts.
[6] Seoane-Vazquez E, Rodriguez-Monguio R, Szeinbach SL, et al. Incentives for orphan drug research and development in the United States[J]. Orphanet J Rare Dis, 2008, 12: 3-33.
[7] Patel S, Miller Needleman KI. FDA's Office of Orphan Products Development: providing incentives to promote the development of products for rare diseases[J]. J Pharmacokinet Pharmacodyn, 2019, 46: 387-393. doi: 10.1007/s10928-019-09645-4
[8] Food and Drug Administration. Search Orphan Drug Designations and Approvals[EB/OL]. (2017-10-24)[2022-05-18]. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/index.cfm#Search_param_field#Search_param_field.
[9] European Medicines Agency. Orphan designation: Overview[EB/OL]. (2021-01-01)[2022-05-18]. https://www.ema.europa.eu/en/human-regulatory/overview/orph-an-designation-overview.
[10] European Medicines Agency. Legal framework: orphan designation[EB/OL]. (2021-01-01)[2022-05-18]. https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation/legal-framework-orphan-designation#regulation-(ec)-no-141/2000-(the-orphan-regulation)-section.
[11] European Medicines Agency. Committee for Orphan Medicinal Products (COMP)[EB/OL]. (2021-10-11)[2022-05-18]. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/comp-rules-procedure_en.pdf.
[12] European Medicines Agency. Applying for orphan designa-tion[EB/OL]. (2021-01-01)[2022-05-08]. https://www.ema.europa.eu/en/human-regulatory/research-develop-ment/orphan-designation/applying-orphan-designation.
[13] European Commission. Community Register of orphan medicinal products[EB/OL]. (2022-05-08)[2022-05-18]. https://ec.europa.eu/health/documents/community-register/html/reg_od_act.htm?sort=n.
[14] 厚生劳动省. 稀有疾病药品、罕见疾病医疗器械、罕见疾病再生医学等产品的认定制度概述[EB/OL]. (2022-05-08)[2022-05-18]. https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/0000068484.html. [15] Sakushima K, Takeda H, Aoi Y. Orphan drug designation and development in Japan: 25 years of experience and assessment[J]. Nat Rev Drug Discov, 2021, 20: 893-894. doi: 10.1038/d41573-021-00045-3
[16] Song P, Gao J, Inagaki Y, et al. Rare diseases, orphan drugs, and their regulation in Asia: current status and future perspectives[J]. Intractable Rare Dis Res, 2012, 1: 3-9.
[17] 丁志琛, 韦冠, 丁锦希. 日本罕用药制度及其对中国的启示——基于对日本罕用药可及性的评价[J]. 中国药科大学学报, 2014, 45: 118-124. https://www.cnki.com.cn/Article/CJFDTOTAL-ZGYD201401028.htm [18] Pharmaceuticals and Medical Devices Agency. List of Approved Products[EB/OL]. (2022-05-08)[2022-05-18]. https://www.nibiohn.go.jp/nibio/part/promote/files/orphan%20drug%20E.pdf.
[19] Biglia LV, Mendes SJ, Lima TM, et al. Incorporation of drugs for rare diseases in Brazil: is it possible to have full access to these patients?[J]. Cien Saude Colet, 2021, 26: 5547-5560. doi: 10.1590/1413-812320212611.26722020
[20] Mayrides M, Ruiz de Castilla EM, Szelepski S. A civil society view of rare disease public policy in six Latin American countries[J]. Orphanet J Rare Dis, 2020, 15: 60. doi: 10.1186/s13023-020-1314-z
[21] Choudhury MC, Saberwal G. The work, goals, challenges, achievements, and recommendations of orphan medicinal product organizations in India: an interview-based study[J]. Orphanet J Rare Dis, 2019, 14: 241. doi: 10.1186/s13023-019-1224-0
[22] Khosla N, Valdez R. A compilation of national plans, policies and government actions for rare diseases in 23 countries[J]. Intractable Rare Dis Res, 2018, 7: 213-222. doi: 10.5582/irdr.2018.01085
[23] 牛叔文, 李真, 梁曼. 人文发展的多维测度及其政策启示[J]. 中国软科学, 2021, (05): 99-110. https://www.cnki.com.cn/Article/CJFDTOTAL-ZGRK202105010.htm [24] 国家药品监督管理局. 国家药监局综合司公开征求《中华人民共和国药品管理法实施条例(修订草案征求意见稿)》意见[EB/OL]. (2022-05-09)[2022-05-18]. https://www.nmpa.gov.cn/xxgk/zhqyj/zhqyjyp/20220509222233134.html. -
期刊类型引用(3)
1. 曲春燕,贾欣玥,金梦轲,史泱,尹梦雅,崔芷君,梁爱民. 儿童语言障碍门诊中84例听力障碍病例特点分析. 中国听力语言康复科学杂志. 2024(06): 561-566 .  百度学术
百度学术
2. 巢薇,覃凤娴,刘小勇,黄以琛. 新生儿耳聋基因诊断技术进展研究. 内科. 2023(02): 149-153 . 百度学术
3. 雷一波,孙淑萍,毛璐,许红恩,汤文学,潘昭宇,卢伟. LARS2和 HARS2基因致病变异所致的Perrault综合征分析. 中华耳鼻咽喉头颈外科杂志. 2023(12): 1191-1197 . 百度学术
其他类型引用(0)
计量
- 文章访问数: 356
- HTML全文浏览量: 116
- PDF下载量: 98
- 被引次数: 3
