 作者投稿
作者投稿 专家审稿
专家审稿 编辑办公
编辑办公 主编办公
主编办公
Mutation Analysis of the CYP4F22 Gene in a Family with Autosomal Recessive Congenital Ichthyosis
-
摘要:
先天性常染色体隐性遗传性鱼鳞病(ARCI)是一种罕见的遗传性角化性皮肤病,表现为全身皮肤干燥脱屑。本文通过二代测序技术,对一例临床诊断为ARCI的患儿进行基因检测,应用Sanger测序对先证者和父母的DNA进行双向验证。结果表明先证者CYP4F22基因存在c.235G>T和c.641delG复合杂合突变,通过变异位点解读指南评估分类为致病性变异,父母分别为两个变异的携带者。CYP4F22基因复合杂合突变是该先证者的致病突变,并且c.235G>T和c.641delG均为文献及数据库未报道的新变异,变异的检出为该家系的遗传咨询奠定了基础。
-
关键词:
- 先天性常染色体隐性遗传性鱼鳞病 /
- CYP4F22基因 /
- 二代测序 /
- 致病性变异 /
- 基因型与表型
Abstract:Autosomal recessive congenital ichthyosis (ARCI) is a rare hereditary cornification disorder presented with abnormal skin scaling. In this paper, we used next-generation sequencing to determine the variants in a Chinese ARCI patient. We used sanger sequencing to verify bidirectionally the DNA from the proband and her parents. Results showes that two compound heterozygous variants (c.235G > T and c.641delG) in CYP4F22 gene, and both of the mutations are novel. The parents were heterozygous carriers. The two variants are classified as pathogenic variants based on interpretation guidelines. The compound heterozygous mutations in CYP4F22 gene were the causative mutations responsible for ARCI in proband.
-
先天性常染色体隐性遗传性鱼鳞病(autosomal recessive congenital ichthyosis, ARCI, OMIM242300)是一种罕见的非综合征性鱼鳞病,具有遗传和表型异质性,表现为全身皮肤干燥及脱屑。主要包括三种亚型:板层状鱼鳞病(lamellar ichthyosis, LI)、先天性鱼鳞病样红皮病(congenital ichthyosiform erythroderma, CIE)、丑角样鱼鳞病(harlequin ichthyosis, HI),此外还包括以下几种少见亚型:自我改善型火棉胶鱼鳞病(self-improving collodion ichthyosis, SICI)、泳衣鱼鳞病(bathing suit ichthyosis, BSI)等。
目前已证实的ARCI致病基因有14种:TGM1、ALOX12B、ALOXE3、ABCA12、CYP4F22、NIPAL4、SDR9C7、LIPN、CERS3、PNPLA1、ST14、CASP14、 SULT2B1和ABCI7,其中CYP4F22较少,约占10%[1]。这些基因编码的蛋白质通过脂质代谢的各种途径参与影响皮肤屏障的正常功能[2]。CYP4F22基因变异在意大利、伊朗、西班牙及其他一些地中海国家均有报道[1, 3-4]。中国相关报道较少,仅有2例[5]。本研究中对1例ARCI患儿进行二代测序,鉴定出CYP4F22基因的2种未见文献报道的新变异,并进一步探讨CYP4F22基因突变与临床表型之间的联系,为该病的诊断和遗传咨询提供依据。
1. 临床资料
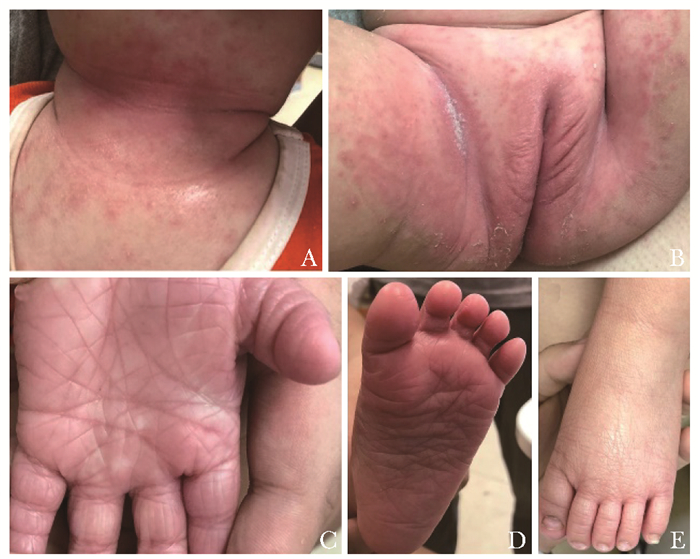
患儿女性,2岁,出生时全身皮肤发红,出现火棉胶样改变,随后渐脱屑。查体:颈部、腋下、外阴、双侧腹股沟散在红色斑片,上覆白色鳞屑,全身皮肤干燥脱屑,双手掌、足跖片状角化过度性红斑,其上皮肤纹理加深。患儿毛发、牙齿、指甲正常;生长发育正常;无睑外翻、唇外翻等(图 1)。先证者父母表型正常,非近亲婚配,否认家族中类似疾病史。临床诊断:ARCI。
![]() 图 1 先天性常染色体隐性遗传性鱼鳞病患儿临床特征A、B.颈部、外阴、双侧腹股沟散在红色斑片,上覆细小白色鳞屑;C、D、E.双手足掌跖角化过度性红斑,皮纹粗糙Figure 1. Clinical features of patient with autosomal recessive congenital ichthyosis
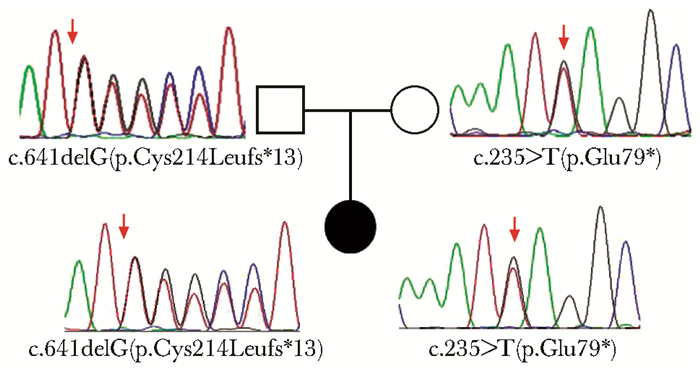
图 1 先天性常染色体隐性遗传性鱼鳞病患儿临床特征A、B.颈部、外阴、双侧腹股沟散在红色斑片,上覆细小白色鳞屑;C、D、E.双手足掌跖角化过度性红斑,皮纹粗糙Figure 1. Clinical features of patient with autosomal recessive congenital ichthyosis采集患儿及其父母外周血各2 mL,使用DNA提取试剂盒(北京天根生化科技有限公司)提取基因组DNA。本研究征得监护人同意并签署知情同意书。采用GenCap遗传性皮肤疾病基因捕获探针对基因组目的基因进行捕获并构建文库,该探针覆盖基因组中726个皮肤病相关的致病基因序列(长度约2.466 Mb),构建好的文库在美国Illumina公司的Novoseq测序仪上进行高通量测序。通过美国医学遗传学会/分子病理学协会(American College of Medical Genetics and Genomics and the Association for Molecular Patho-logy, ACMG/AMP)的指南对变异进行致病性分类[6]。通过Primer Premier 5软件在CYP4F22基因检测到的致病突变位点上下游设计引物,用Sanger测序进行验证。发现先证者CYP4F22基因存在c.235G>T(p.Glu79*)和c.641delG(p.Cys214Leufs*13) 两个变异。经家系验证分析,c.235G>T(p.Glu79*) 来自其母亲,该变异位于4号外显子,导致79位氨基酸谷氨酸突变,提前出现终止密码子;c.641delG(p.Cys214Leufs*13)来自其父亲,该变异位于7号外显子,导致214位半胱氨酸突变为亮氨酸,并在其后13位提前出现终止密码子(图 2)。这两个变异预计会通过“无义介导的mRNA衰减”机制降解或产生截短蛋白,因而突变等位基因活性大部分丧失。两种变异在dbSNP数据库、千人基因组数据库以及gnomAD数据库中均未见收录,提示等位基因变异人群频率极低,为未见文献及数据库报道的新变异。综上所述,根据ACMG指南,将CYP4F22基因c.235G>T(p.Glu79*)和c.641delG(p.Cys214Leufs*13)变异分级为致病性变异。
![]() 图 2 先证者家系图及Sanger测序突变位点用红色箭头表示Figure 2. Pedigree chart of peroband and sanger sequencing results
图 2 先证者家系图及Sanger测序突变位点用红色箭头表示Figure 2. Pedigree chart of peroband and sanger sequencing results2. 讨论
CYP4F22基因既往称作FLJ39501基因,定位于染色体19p13.12,包含14个外显子。CYP4F22基因编码在表皮中高度表达的ω-羟化酶,它对酰基神经酰胺的合成至关重要,该基因致病性变异会导致表皮酰基神经酰胺的生成减少,影响皮肤屏障的形成及其正常功能[7-8]。2006年Lefevre等[4]学者对12个近亲婚配家庭中的ARCI患者进行连锁分析,首次确定了CYP4F22基因发生变异,会导致LI和掌跖部位纹理粗糙的表现。2020年,一项对92例西班牙ARCI患者的研究发现,其中8个家系携带CYP4F22基因突变,这些患者最常见的症状为出生时存在火棉胶膜、掌跖区域纹理加深,并且鉴定出的c.1303C>T(p.His435Tyr)突变被证实为西班牙人群中的创始人突变[9]。目前已报道的可引起ARCI的CYP4F22基因变异仅55个,其中错义变异38个、移码变异11个、剪切位点变异6个。本研究中,c.235G>T和c.641delG均未见文献及数据库报道。CYP4F22基因突变频率在不同人群中存在差异。在伊朗和意大利人群中占2%~6%[3, 10],而在可能存在创始人突变效应的人群中,频率可达到10%~12%[11-12]。
ARCI的致病基因较多,与各临床亚型的关系尚未完全清楚。除比较严重的HI亚型主要由ABCA12基因功能丧失性突变引起外,包括CYP4F22基因在内的大多数已知的ARCI致病基因的变异可能导致患者表现为较轻的LI、CIE或SICI亚型[9, 11, 13]。一般来说,具有CYP4F22突变的患者在出生时往往更易表现出红皮病的状态,之后大多表现为LI或CIE表型。由于ARCI不同亚型患者表型之间的重叠性,以及随病程发展表型的不同变化,仅根据临床特征进行分型诊断十分困难。因此,ARCI患者进行分子遗传学分析有助于患者确诊、治疗及预后评估,也是家庭中的基因诊断与产前诊断最可靠的方法。
在ARCI患者中已有关于皮肤肿瘤的报道,多与皮肤屏障受损导致的皮肤慢性炎症有关[14-15]。Hotz等[11]学者在一项包括54个携带CYP4F22突变家系的队列研究中发现,2例患者自40岁以后出现高分化鳞状细胞癌或者结节样基底细胞癌,这2例患者的共同点是:都诊断为成人CIE,且四肢多发红斑丘疹。由此Hotz等学者认为伴有多发性红斑丘疹的CYP4F22基因突变患者出现皮肤恶性肿瘤的风险可能更高,但CYP4F22基因突变和皮肤恶性肿瘤的直接相关性还有待进一步研究。本研究中患者为2岁儿童,未出现四肢红斑丘疹或皮肤肿物,但仍需对其进行长期随访,警惕皮肤恶性肿瘤发生的可能,必要时可进行相关检查。
迄今为止,在携带CYP4F22基因致病变异的ARCI患者中,基因型与表型关系尚不十分明确。CYP4F22蛋白由两个底物结合区(substrate-binding regions, SBRs)组成。有研究表明,携带一个或两个影响SBR区域的CYP4F22截短突变的患者在出生时往往会有火棉胶样改变。2016年,Feng等[5]报道了2例携带CYP4F22纯合错义突变的中国患者,其中携带位于SBR区域纯合突变的患者出生时为火棉胶婴儿,而在SBR区域之外的纯合突变的患者在出生时表型相对温和,无火棉胶膜,因此,推测影响SBR区域的错义突变也可能与出生时火棉胶膜的发育有关。本患者为CYP4F22基因截短突变的复合杂合子,这两个截短突变皆会影响SBR区域,患者在出生时也出现火棉胶样改变,符合之前学者的结论。但CYP4F22基因突变位置或类型与表型严重程度之间仍缺乏关联性,需要进一步研究以明确。Fioretti曾报道过一例携带CYP4F22基因两个截短突变的复合杂合患者(c.76_85del和Ex.3del),该患者出生时伴火棉胶膜,此外还有少汗、肘膝部严重角化过度的表现[16]。与之相比,本研究中先证者虽携带两个截短变异,但明显表现出较轻的表型。
ARCI应与具有鱼鳞病样表现的其他疾病相鉴别,如Netherton综合征、角膜炎-鱼鳞病-耳聋综合征(keratitis ichthyosis deafness syn- drome, KID syndrome)、Sjogren-Larsson综合征(Sjogren-Larsson syndrome, SLS) 等。Netherton综合征是一种罕见的常染色体隐性遗传病,由SPINK5基因变异引起,临床表现除旋涡状(迂回线状)鱼鳞病外,还有发干异常(竹节状脆发)、特异性体质包括IgE升高、荨麻疹等。婴儿期危及生命的并发症包括体温和电解质失衡、反复感染和败血症。KID综合征是一种多由GJB2基因变异引起常染色体显性遗传性疾病,以血管性角膜炎、先天性鱼鳞病、掌跖角化过度和中度至重度感音神经性耳聋为特征。SLS是一种常染色体隐性遗传性疾病,多于儿童早期发病,由ALDH3A2基因突变引起,其特征为鱼鳞病、智力低下、痉挛性下肢轻瘫、黄斑营养不良等。此类疾病的诊断及鉴别诊断应基于皮肤表现、个人和家族史以及皮肤外表现等方面的综合评估,基因检测则有助于确诊和遗传咨询。
对于患ARCI的新生儿来说,需确保环境潮湿、预防感染,对新生儿的正确监护有利于改善严重先天性鱼鳞病的结局。随着患儿年龄的增长,可以通过增加沐浴时间改善症状,使用保湿剂保持皮肤滋润,外用角质剥脱剂如水杨酸、尿素等促进鳞屑剥脱。对于皮肤受累严重的患者,也可以系统性口服维A酸类药物治疗,在治疗期间应注意药物剂量、使用时间,并定期进行相关实验室检查,避免使用该类药物出现的毒副作用,如肝毒性或骨损害等[17]。
综上所述,本研究报道了1例罕见的由CYP4F22基因突变导致的ARCI患者,鉴定出两个新变异,扩展了CYP4F22基因突变频谱,对进一步明确CYP4F22基因突变与ARCI临床表型之间的关系提供了有意义的线索,并且为患者及其家庭的遗传咨询奠定了基础。
作者贡献:论文选题与设计:徐哲;论文整合与修订:徐哲,张樱子;撰稿:张樱子;数据整理与分析:张樱子,石海涛,刘腾,赵洋。利益冲突:所有作者均声明不存在利益冲突。 -
![]()
图 1 先天性常染色体隐性遗传性鱼鳞病患儿临床特征
A、B.颈部、外阴、双侧腹股沟散在红色斑片,上覆细小白色鳞屑;C、D、E.双手足掌跖角化过度性红斑,皮纹粗糙
Figure 1. Clinical features of patient with autosomal recessive congenital ichthyosis
-
[1] Pigg MH, Bygum A, Ganemo A, et al. Spectrum of autosomal recessive congenital ichthyosis in scandinavia: clinical characteristics and novel and recurrent mutations in 132 patients[J]. Acta Derm Venereol 2016, 96: 932-937. doi: 10.2340/00015555-2418
[2] Vahlquist A, Fischer J, Torma H. Inherited nonsyndromic ichthyoses: an update on pathophysiology, diagnosis and treatment[J]. Am J Clin Dermatol, 2018, 19: 51-66. doi: 10.1007/s40257-017-0313-x
[3] Youssefian L, Vahidnezhad H, Saeidian AH, et al. Autosomal recessive congenital ichthyosis: Genomic landscape and phenotypic spectrum in a cohort of 125 consanguineous families[J]. Hum Mutat, 2019, 40: 288-298. doi: 10.1002/humu.23695
[4] Lefevre C, Bouadjar B, Ferrand V, et al. Mutations in a new cytochrome P450 gene in lamellar ichthyosis type 3[J]. Hum Mol Genet, 2006, 15: 767-776. doi: 10.1093/hmg/ddi491
[5] Feng C, Wang H, Lee M, et al. Two missense mutations in CYP4F22 in autosomal recessive congenital ichthyosis[J]. Clin Exp Dermatol, 2017, 42: 98-100. doi: 10.1111/ced.12988
[6] Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17: 405-424. doi: 10.1038/gim.2015.30
[7] Behne M, Uchida Y, Seki T, et al. Omega-hydroxycera-mides are required for corneocyte lipid envelope (CLE) formation and normal epidermal permeability barrier function[J]. J Invest Dermatol, 2000, 114: 185-192. doi: 10.1046/j.1523-1747.2000.00846.x
[8] Ohno Y, Nakamichi S, Ohkuni A, et al. Essential role of the cytochrome P450 CYP4F22 in the production of acylceramide, the key lipid for skin permeability barrier formation[J]. Proc Natl Acad Sci USA, 2015, 112: 7707-7712. doi: 10.1073/pnas.1503491112
[9] Esperon-Moldes U, Ginarte-Val M, Rodriguez-Pazos L, et al. Novel CYP4F22 mutations associated with autosomal recessive congenital ichthyosis (ARCI). Study of the CYP4F22 c. 1303C > T founder mutation[J]. PLoS One, 2020, 15: e0229025. doi: 10.1371/journal.pone.0229025
[10] Diociaiuti A, El Hachem M, Pisaneschi E, et al. Role of molecular testing in the multidisciplinary diagnostic approach of ichthyosis[J]. Orphanet J Care Dis, 2016, 11: 4. doi: 10.1186/s13023-016-0384-4
[11] Hotz A, Bourrat E, Kusel J, et al. Mutation update for CYP4F22 variants associated with autosomal recessive congenital ichthyosis[J]. Hum Mutat, 2018, 39: 1305-1313. doi: 10.1002/humu.23594
[12] Buckova H, Noskova H, Borska R, et al. Autosomal recessive congenital ichthyoses in the Czech Republic[J]. Br J Dermatol, 2016, 174: 405-407. doi: 10.1111/bjd.13918
[13] Takeichi T, Akiyama M. Inherited ichthyosis: Non-syndromic forms[J]. J Dermatol, 2016, 43: 242-251. doi: 10.1111/1346-8138.13243
[14] Natsuga K, Akiyama M, Shimizu H. Malignant skin tumours in patients with inherited ichthyosis[J]. Br J Dermatol, 2011, 165: 263-268. doi: 10.1111/j.1365-2133.2011.10381.x
[15] McKenzie S, Arzeno J, Lonowski S, et al. Increased melanocytic nevi and lentigines in two patients with harlequin ichthyosis[J]. Pediatr Dermatol, 2020, 37: 192-195. doi: 10.1111/pde.14066
[16] Fioretti T, Auricchio L, Piccirillo A, et al. Multi-gene next-generation sequencing for molecular diagnosis of autosomal recessive congenital ichthyosis: A genotype-phenotype study of four italian patients[J]. Diagnostics(Pasel), 2020, 10: 995.
[17] Digiovanna JJ, Mauro T, Milstone LM, et al. Systemic retinoids in the management of ichthyoses and related skin types[J]. Dermatol Ther, 2013, 26: 26-38. doi: 10.1111/j.1529-8019.2012.01527.x
-
期刊类型引用(1)
1. 韩春雨,韩建文. ALOXE3基因突变致常染色体隐性先天性鱼鳞病一例. 中国麻风皮肤病杂志. 2025(01): 9-14 .  百度学术
百度学术
其他类型引用(0)
 下载:
下载:
计量
- 文章访问数: 266
- HTML全文浏览量: 94
- PDF下载量: 51
- 被引次数: 1


