 作者投稿
作者投稿 专家审稿
专家审稿 编辑办公
编辑办公 主编办公
主编办公
-
摘要:
一例15岁青少年女性,因间断发热就诊,由此发现巨脾、继发脾功能亢进、视网膜色素变性,以及多种外胚层发育不良的表现等多系统病症。追问病史,挖掘复杂的既往史,包括反复呼吸道感染、夜间视物不清、牙齿和甲床发育不良等。经北京协和医院儿科风湿免疫专业组的经验分析,疑诊为一种近年新认识的罕见病——ROSAH综合征,最终得到基因测序结果的证实。在生物制剂肿瘤坏死因子抑制剂治疗过程中,该患者再次反复发热,伴炎症指标升高,且新出现头痛。罕见病多学科会诊团队为指导患者综合治疗、改善患者生活质量展开讨论,指导治疗。
Abstract:A 15-year-old female was referred to the hospital with intermittent fever, where multiple systemic abnormalities were found, such as splenomegaly, secondary hypersplenism, retinitis pigmentosa, and ectodermal dysplasia. Medical history revealed that she had suffered recurrent respiratory infections, blurred vision at night, and dysplasia of teeth and nail beds since childhood. Then she was suspected to be experiencing ROSAH syndrome, a rare disease newly recognized in recent years, which was finally confirmed by gene sequencing results. During a course of treatment with tumor necrosis factor inhibitors, recurrent fever with elevated inflammatory markers reappeared, and the child developed headaches. To guide the comprehensive treatment and improve the patient's quality of life, the multidisciplinary team in Peking Union Medical College Hospital discussed together and directed the following treatment.
-
Keywords:
- splenomegaly /
- fundus lesions /
- ectodermal dysplasia /
- headache /
- tumor necrosis factor /
- autoinflammatory diseases
-
1. 病例简介
患者女性,15岁,因“间断发热、发现肝脾肿大2年余,头痛7月”于2021年8月15日在北京协和医院进行罕见病多学科讨论。
1.1 现病史
2018年12月起患者无诱因间断发热,体温37.5 ℃~ 38.2 ℃,每月2~3次,无明显周期性,无伴随症状,未诊治。2019年2月再次发热,性质同前,于当地医院查血WBC 1.94×109/L↓,NEUT# 1.01×109/L(52%),PLT 98×109 /L↓,HGB 83 g/L↓;Ret 4.1%↑。骨髓涂片:增生性贫血,细胞内铁减低。骨髓活检:骨髓增生性贫血病理改变。病原学以及自身抗体检查(-)。腹部超声:肝脾大,右肝斜径14.3 cm,脾厚8.3 cm、长径26.0 cm;脾内实性占位(血管瘤可能)。未明确诊断,建议上级医院就诊。
2019年3月为进一步诊治就诊于北京协和医院血液科,查血WBC 2.4×109/L↓,NEUT# 1.86×109/L(77.5%)↓,PLT 81×109/L↓,HGB 86 g/L↓。铁四项提示缺铁性贫血。骨髓涂片:增生活跃,粒=47.5%,红=46.5%,粒∶红=1.02。粒系各阶段比例及形态大致正常;红系中、晚幼红细胞比例增高,形态大致正常,红细胞大小不等,部分形态不规则,中心淡染区扩大;淋巴细胞比例减低,形态正常;单核细胞比例形态正常;巨核细胞及血小板不少;未见其他异常细胞及寄生虫;铁染色未见环形铁粒幼红细胞。全身PET/CT:肝脏、脾脏显著增大,全身骨髓代谢略高,考虑发热后继发改变可能性大,余未见明显异常。考虑“脾亢”,建议脾脏切除以及儿科就诊。
2019年5月于我院儿科住院,追问既往史:7月龄至1.5岁反复上呼吸道感染,约每月1次;2岁左右发现双眼内斜视,未治疗自行缓解;9岁始夜间视物不清;半岁出牙,1岁4月乳牙多发“龋齿”,恒牙刚萌出正常,很快颜色发黄,龋齿,治疗后仍反复,影像学提示牙根浅,上颌骨不发育;11岁始手指甲面凹凸不平。查体:身高156.5 cm(P25,P50),体重44.3 kg(P25,P50),发育正常,贫血貌,无皮疹,多发龋齿,心肺查体正常,腹软,肝肋下3 cm、剑下未及,脾肋下10 cm,Ⅰ线16 cm,Ⅱ线18 cm,Ⅲ线3 cm。完善检查:血WBC 2.05×109/L↓,HGB 81 g/L↓,PLT 85×109/L↓。尿便常规、肝肾功、血脂正常。Fe 48 μg/dL↓,TRF 2.59 g/L,TIBC 330 μg/dL,IS 14.5%↓,TS 13.1%↓,Fer 13 ng/mL;叶酸和维生素B12正常。炎症指标:CRP 12~40 mg/L↑,ESR 14~ 33 mm/h↑。细胞因子:IL-6 5.3 pg/mL(<5.9 pg/mL),IL-8 73 pg/mL(<62 pg/mL)↑,IL-10 5.0 pg/mL (<9.1 pg/mL),TNF-α 47.2 pg/mL(<8.1 pg/mL)↑。【病原学】ASO 1449 IU/mL↑,余细菌、非典型病原、结核、病毒及真菌(-)。【免疫指标】IgG 16.16 g/L,IgA 1.00 g/L,IgM 1.26 g/L。血清IgG亚类:IgG1 13 300 mg/L,IgG2 3640 mg/L,IgG3 1550 mg/L,IgG4 479 mg/L。C3 1.185 g/L,C4 0.168 g/L。IgD固定电泳(-),IgE正常。
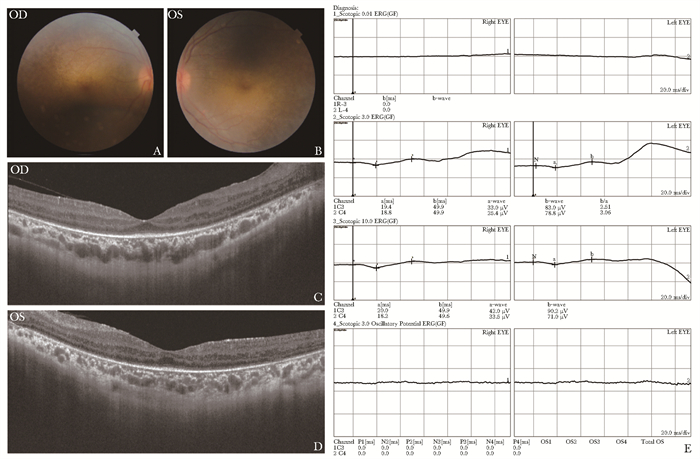
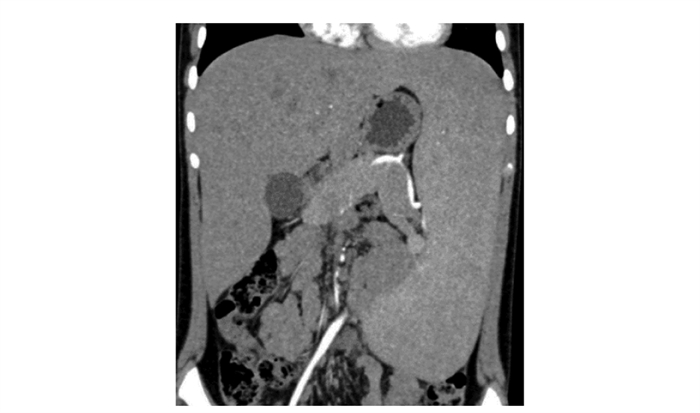
淋巴细胞比例:T% 80.3%,T4% 44.9%,T8% 29.5%,NK% 10.7%,B% 6.4%。RF、ANA3项、ENA(-)。【内分泌】甲功、性激素、ACTH、皮质醇正常。β-CTX 1.68 ng/mL↑,T-25OHD 6.5 ng/mL↓。【遗传代谢】血氨、乳酸正常;β-葡糖苷酶及鞘磷脂酶正常。【影像学】腹部CT增强+CTA:肝脾增大(图 1),脾脏多发不均匀低强化影;门静脉近汇合处增粗,脾静脉增粗迂曲。上消化道造影:胃黏膜粗糙、紊乱。腹部血管及心脏超声:未见异常。【其他】眼科会诊:根据OCT、VF、ERG5项结果(图 2),考虑双眼视网膜色素变性。耳鼻喉科会诊:纯音测听+声导抗正常。皮肤发汗试验阳性。
![]() 图 2 患者的眼科检查结果A、B.视网膜色素紊乱,视盘水肿;C、D.视网膜椭圆体带破坏;E.视网膜电图示各振幅均重度降低Figure 2. Results of the patient's ocular examination
图 2 患者的眼科检查结果A、B.视网膜色素紊乱,视盘水肿;C、D.视网膜椭圆体带破坏;E.视网膜电图示各振幅均重度降低Figure 2. Results of the patient's ocular examination因患儿症状累及肝脾、视网膜、牙齿及指甲等多个系统,不除外遗传代谢性疾病、外胚层发育不良及原发性免疫缺陷,予完善全外显子基因测序。治疗上,暂予补充维生素D、铁剂及叶黄素等对症治疗。
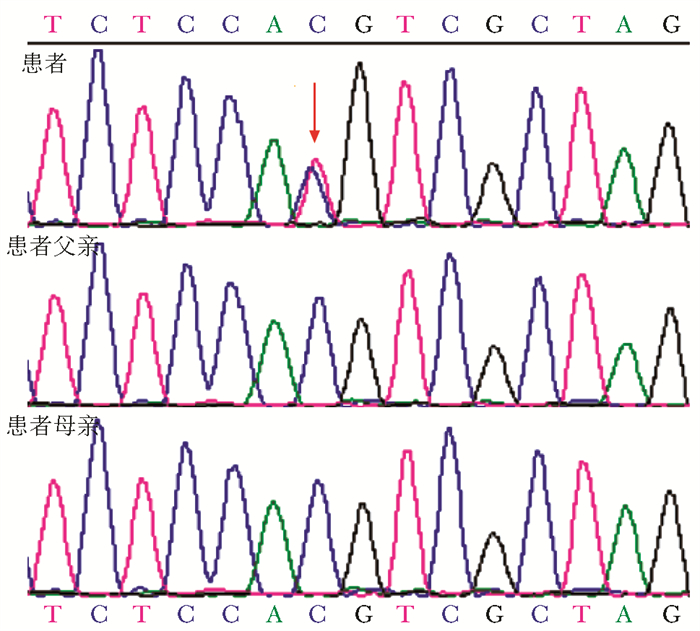
2019年7月我院分析全外显子测序数据,发现ALPK1新生杂合突变(NM_ 001253884):c.476c>T,p.T159M(图 3),对应临床表型为ROSAH综合征(视网膜病变、视盘水肿、脾大、无汗、偏头痛综合征,常染色体显性遗传)。经查阅文献及儿科风湿免疫专业组查房,诊断ROSAH综合征明确,并根据该综合征致病机制以肿瘤坏死因子(tumor necrosis factor,TNF)炎症通路异常激活为主,建议予TNF抑制剂治疗。
![]() 图 3 患者及其父母的Sanger测序结果突变位点用红色箭头表示Figure 3. The Sanger sequencing results of the patient and her parents
图 3 患者及其父母的Sanger测序结果突变位点用红色箭头表示Figure 3. The Sanger sequencing results of the patient and her parents排除禁忌后于2019年11月1日至2020年7月行1~7程英夫利昔单抗280 mg/次治疗[体重波动于48~50 kg之间,合5.6~5.8 mg/(kg·次)],分别在0、2、6周,此后每8周应用1次。患儿1~4次治疗期间监测炎症指标较前下降,肝脾逐渐减小,血三系仍低,自5程英夫利昔单抗后(2020年4月)再次间断发热,每周发热1日,体温最高38 ℃,体温可自行降至正常,不伴其他不适,继续英夫利昔单抗治疗。2020年9月第8程英夫利昔单抗治疗前返院评估:血三系仍减低;CRP 76 mg/L↑,ESR 38 mm/h↑;IL-6 6.1 pg/mL↑,TNF-α 257.0 pg/mL↑。腹部超声示肝脾较前增大(右肝斜径13→14.1 cm,脾厚5.3→5.7 cm,脾长径18.8→20.5 cm)。头颅CT未见异常。眼科较前无明显进展。儿科风湿免疫专业组考虑不除外注射用英夫利昔单抗(商品名:类克)失应答,留取血标本检测血药浓度和单抗抗体后,将本次英夫利昔单抗加量至400 mg/次[体重50 kg,合8 mg/(kg·次)]。于2020年9月28日输注第8程英夫利昔单抗,后结果回示血药浓度<0.4 μg/mL↓,英夫利昔单抗抗体74 ng/mL↑。结合脾脏、血三系、视网膜病变未见明显缓解,且英夫利昔单抗继发性失应答,调整为阿达木单抗治疗,2020月12月1日第1次阿达木单抗40 mg治疗,过程顺利。
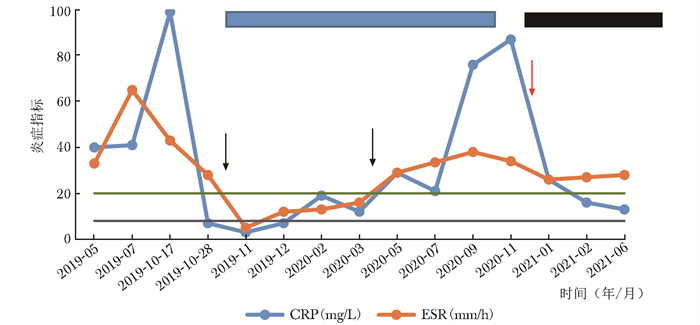
数天后开始无诱因反复头顶部、眼眶及眼球疼痛,无呕吐,每周1~2次,每次持续半日左右,间断发热同前,发热时头痛加重,当地头CT平扫未见异常,眼科检查后诊断“虹膜炎”,当地对症治疗后无明显改善,后家长认为阿达木单抗副作用,自行停药。2021年1月底因头痛返院评估:血三系、炎症指标、肝脾较前无明显变化。头MRI+磁敏感加权成像(susceptibility weighted imaging, SWI):垂体饱满;双侧上颌窦黏膜增厚。经颅多普勒血管超声、脑电图正常。拒绝腰穿。眼科会诊:眼压正常,眼底无水肿,眼部表现符合广泛的杆-锥细胞受损,视野较前进一步缩小,视网膜电图示双眼α、β波振幅重度降低。儿科风湿免疫专业组查房考虑原发病相关偏头痛,建议继续阿达木单抗治疗。后偶有头痛、发热,发热可自行恢复,继续应用至2021年5月21日,因再次头痛自行停药,并予甘露醇治疗,头痛明显好转。2021年6月28日返院评估:头MRI+SWI未见明显异常。余系统评估无明显变化。建议规律阿达木单抗治疗。后每月发热2次,偶有头痛,情况同前。炎症指标与症状随治疗变化见图 4。
![]() 图 4 患者炎症指标与症状随治疗变化情况蓝色方块为英夫利昔单抗治疗期间,黑色方块为阿达木单抗治疗期间;两个黑色箭头之间无发热,之外均有反复发热;红色箭头为头痛开始的时间点Figure 4. Inflammatory indicators and symptoms change with treatment
图 4 患者炎症指标与症状随治疗变化情况蓝色方块为英夫利昔单抗治疗期间,黑色方块为阿达木单抗治疗期间;两个黑色箭头之间无发热,之外均有反复发热;红色箭头为头痛开始的时间点Figure 4. Inflammatory indicators and symptoms change with treatment起病以来,患儿精神可,饮食及睡眠正常,二便如常,体重无减轻。
1.2 既往史、个人史及家族史
7月龄至1.5岁反复上呼吸道感染,约每月1次。2岁左右发现双眼内斜视,未治疗自行缓解。9岁始夜间视物不清。半岁出牙,1岁4月乳牙多发“龋齿”,恒牙刚萌出正常,很快颜色发黄,龋齿,治疗后仍反复,影像学提示牙齿牙根浅,上颌骨不发育。11岁始手指甲面凹凸不平。
患儿系第4胎第1产儿(母亲曾因社会因素或不明原因胚胎停育流产),足月剖宫产娩出,无缺氧窒息史,生后母乳喂养,余无殊。体格、运动、智力、语言发育似同龄儿。按计划接种疫苗,无不良反应。
家族史无特殊。
1.3 查体
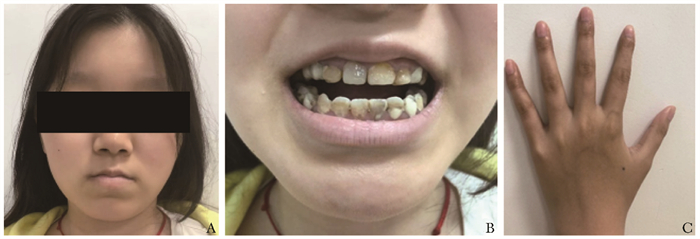
身高164 cm(P50,P75),体重55 kg(P50,P75)。发育正常,营养良好,贫血面容。四肢皮肤干燥,未见脱屑。头发、眉毛、睫毛稀疏。浅表淋巴结未及肿大。口腔内多发龋齿,牙釉质发黄。肝肋下约4 cm,剑下3 cm,质中。脾肿大,Ⅰ线约13 cm,Ⅱ线约16 cm,Ⅲ线0 cm。余腹部、心肺查体大致正常,四肢关节活动自如(图 5)。双手杵状指(±),指甲凹凸不平,双足踇趾外翻。
![]() 图 5 患者颜面部、口腔及手指变化情况A.眉毛、头发稀疏、细软,贫血貌;B.龋齿、牙釉质发黄;C.杵状指Figure 5. The changes of patient's face, mouth and fingers
图 5 患者颜面部、口腔及手指变化情况A.眉毛、头发稀疏、细软,贫血貌;B.龋齿、牙釉质发黄;C.杵状指Figure 5. The changes of patient's face, mouth and fingers2. 多学科讨论
2.1 儿科马明圣
青少年女性,隐匿起病,慢性病程;主要表现为间断发热、脾大、继发脾亢、眼底病变、头痛;查体发现外胚层发育不良,如牙齿、毛发、指甲发育不良;ESR、CRP及TNF-α等炎症指标升高;基因测序提示与临床表现相符的ALPK1基因已知致病突变;因此诊断ROSAH综合征明确。ROSAH综合征是视网膜病变、视盘水肿、脾大、无汗、偏头痛的英文单词首字母缩写。经过文献复习发现,除上述典型表现外,多数患者均有外胚层发育不良表现。治疗上,多数病例使用大剂量糖皮质激素缓解全身炎症;其他免疫抑制剂,包括秋水仙碱、吗替麦考酚酯、白介素1抑制剂等,虽然也可以缓解全身炎症,但却不能阻止眼底病变进展。另外,针对本病的头痛用非甾体类抗炎药治疗效果差。本患儿选择肿瘤坏死因子抑制剂治疗,病程中曾有好转,但现在控制欠佳,包括再次反复发热、炎症指标升高及新出现头痛。因此,请各科老师指导,是否有更好的治疗手段。另外,患儿脾亢进明显,血三系减低,请外科评估是否考虑脾切除。
2.2 儿科宋红梅
患儿病史2年余,临床主要表现为间断发热、肝脾大、血三系减低,视网膜色素变性,外胚层发育不良;全外显子测序提示ALPK1基因新生已知致病突变,根据以上ROSAH综合征诊断明确;应用TNF抑制剂治疗,曾用药不规律,近7个月出现头痛,分析可能是原发病相关的头痛,或阿达木单抗注射液副作用,也可能是该患儿长期患病就诊相关的非特异性主诉。总体印象上患儿头痛时全身炎症指标升高,提示原发病控制欠佳,且原发病出现发热时头痛加重,考虑原发病相关头痛可能性更大。另患儿原发病出现巨脾、脾功能亢进,血三系减低,神经系统感染、出血等风险高,也会出现头痛,需警惕,请各位专家评估是否有脾切除手术指征,减低出血和感染风险。请各位专家协助综合诊疗。
2.3 眼科睢瑞芳
根据文献报道及临床观察发现该病患者临床表型差异较大,眼部受累表现多种多样,包括葡萄膜炎、视乳头水肿、视网膜水肿或劈裂等,色觉或视野异常,视锥、视杆营养不良等。该患儿存在视乳头水肿、视网膜病变,整体上符合视网膜色素变性的特点。该患儿头痛初期伴有眼痛,后头痛时有发生,但不再伴有眼痛,多次测量眼压正常,监测眼底病变无明显变化,且该综合征本身也常出现头痛,因此考虑该患儿头痛与眼底病变关系不大。针对眼病,治疗后注意随访眼底病变,如果效果不好,可考虑眼睛局部加强治疗。
2.4 心理医学科洪霞
患儿近期出现头痛,应考虑器质性、药物性及心理性因素,患儿系青春期女孩,目前无焦虑及抑郁等心理疾病证据,需侧重器质性病变等原因排查,心理因素方面应注意压力管理、心理支持。
2.5 放射科冯逢
患儿多次头颅影像均未见颅脑占位、出血或感染等器质性病变的征象,腹部影像学显示脾内异常信号,考虑脾梗死可能性大。
2.6 核医学科霍力
患儿PET/CT显示肝脾大,无肿瘤性病变提示,总体表现缺乏特异性。
2.7 基本外科徐强/戴梦华
该患儿脾脏肿大,继发脾功能亢进、血三系减低,但无反复感染、出血等表现,结合血三系水平考虑为轻度脾功能亢进。结合头颅影像学及临床,不考虑头痛为颅内感染或出血所致,无紧急脾切除指征。目前治疗过程中注意监测血三系变化,如脾功能亢进仍不缓解,血小板继续下降,可择期行脾切除术。
3. 多学科讨论后处理
综合多学科专家会诊意见,原发病方面,继续规律应用TNF抑制剂;心理因素方面注意压力管理、心理支持;眼科定期随诊眼底病变;定期监测血三系变化,如规律应用TNF抑制剂治疗,脾功能亢进不缓解,血三系维持欠佳,可考虑外科行脾切除术。
4. 最终诊断
(1) ROSAH综合征(偏头痛,肝大、门静脉增宽,脾大、脾功能亢进、全血细胞减少、脾内低回声,虹膜炎,视网膜色素变性,牙釉质发育不全、龋齿);(2)英夫利昔单抗继发失应答;(3)铁缺乏;(4)双侧足踇趾外翻。
5. 治疗及随访
患儿规律应用TNF抑制剂,间断轻度头痛,家长认为阿达木单抗注射液(商品名:修美乐)疗效不佳,2021年11月26日注射后自行停用。后仍间断发热、头痛,间隔及热峰无明显改变。2022年1—2月再次返院,复查炎症指标仍高,头颅MRI未见异常,腰椎穿刺提示脑脊液压力正常(120 mm H2O),脑脊液常规、生化及病原学未见明显异常。儿科风湿免疫专业组查房后,为控制全身炎症,予泼尼松10 mg/d口服,后患儿发热、头痛有改善,疼痛程度较前减轻,1~2周发作1次,每次持续2~10 h不等,经休息可缓解,基本不影响学习和生活,监测血三系基本同前。
6. 讨论
ROSAH综合征(retinal dystrophy,optic nerve edema,splenomegaly,anhidrosis and migraine headache syndrome,ROSAH)是2019年4月首次被报道的常染色体显性遗传病,致病基因为ALPK1,临床特征是视网膜营养不良、视神经水肿、脾大、无汗和偏头痛[1]。ROSAH综合征患者主要表现为青少年期发病的眼底病变和脾肿大,其他包括头痛、牙齿和甲床发育不良、反复发热、关节炎、皮疹或反复感染等[2-5]。ALPK1基因的p.T159M为热点突变[2-5]。目前对该综合征的治疗及预后知之甚少,已报道的病例最终多采用肿瘤坏死因子抑制剂治疗,效果有待于进一步观察;该病发现时眼底病变多已十分严重,且部分病例诊断时已失明,显著影响患者生活质量。由于认识的局限,及临床表型的重叠,一些患者被误诊为某些多基因自身炎症性疾病,使得诊治工作开展更为延后。该综合征累及多个系统,临床诊治可能涉及多个专科,包括风湿免疫科、血液科、外科、眼科等,综合治疗方案复杂,且需要长期随访,多科共同管理对该病患儿的治疗和预后意义重大。
7. 专家点评
儿科宋红梅
ROSAH是“retinal dystrophy, optic nerve edema, splenomegaly, anhidrosis, and migraine headache”的缩写,中文名称是视网膜营养不良、视神经水肿、脾肿大、无汗、偏头痛,这也是ROSAH综合征的临床特点,其中以儿童期即发现的眼底病变、脾脏肿大伴脾功能亢进最为突出。北京协和医院儿科风湿免疫团队基于长期在原发性免疫缺陷病领域的深耕细作,积攒了相关疾病丰富的诊治经验。本例ROSAH综合征的确诊便是如此。基于诊断、探索治疗,根据致病基因ALPK1的编码蛋白——α激酶1所在的炎症通路,本患儿采用了TNF抑制剂治疗,使其病情得到了一定程度的控制。由于原发性免疫缺陷病临床表现的异质性,治疗过程中每一种新症状的出现均应判断是原发病表现、药物副作用还是其他合并症等,这是一种相对复杂的思辨过程,甚至充分论证后,仍不能得到确切答案。本患儿在TNF抑制剂治疗过程中新出现头痛,与头痛症状同期出现的还有炎症指标升高,发热时头痛加重,均提示原发病控制欠佳,考虑头痛为原发病相关可能性大,经罕见病多学科会诊讨论后建议坚持原发病治疗,并辅助心理支持,使患儿生活质量改善。
作者贡献:论文选题:宋红梅,马明圣;临床诊治:钟林庆,马明圣,宋红梅,睢瑞芳,洪霞,冯逢,霍力,戴梦华,徐强;数据收集:钟林庆;文章撰写与研究设计:钟林庆,马明圣,宋红梅;文章审阅与修改:宋红梅,马明圣。利益冲突:所有作者均声明不存在利益冲突。 -
![]()
图 2 患者的眼科检查结果
A、B.视网膜色素紊乱,视盘水肿;C、D.视网膜椭圆体带破坏;E.视网膜电图示各振幅均重度降低
Figure 2. Results of the patient's ocular examination
![]()
图 3 患者及其父母的Sanger测序结果
突变位点用红色箭头表示
Figure 3. The Sanger sequencing results of the patient and her parents
![]()
图 4 患者炎症指标与症状随治疗变化情况
蓝色方块为英夫利昔单抗治疗期间,黑色方块为阿达木单抗治疗期间;两个黑色箭头之间无发热,之外均有反复发热;红色箭头为头痛开始的时间点
Figure 4. Inflammatory indicators and symptoms change with treatment
-
[1] Williams LB, Javed A, Sabri A, et al. ALPK1 missense pathogenic variant in five families leads to ROSAH syndrome, an ocular multisystem autosomal dominant disorder[J]. Genet Med, 2019, 21: 2103-2115. doi: 10.1038/s41436-019-0476-3
[2] Jamilloux Y, Mathis T, Grunewald O, et al. ALPK1 gene mutations drive autoinflammation with ectodermal dysplasia and progressive vision loss[J]. J Clin Immunol, 2021, 41: 1671-1673. doi: 10.1007/s10875-021-01087-3
[3] Zhong L, Wang J, Wang W, et al. Juvenile onset splenomegaly and oculopathy due to germline mutation in ALPK1[J]. J Clin Immunol, 2020, 40: 350-358. doi: 10.1007/s10875-020-00741-6
[4] Sangiorgi E, Azzarà A, Molinario C, et al. Rare missense variants in the ALPK1 gene may predispose to periodic fever, aphthous stomatitis, pharyngitis and adenitis (PFAPA) syndrome[J]. Eur J Hum Genet, 2019, 27: 1361-1368. doi: 10.1038/s41431-019-0421-6
[5] Hecker J, Letizia M, Loescher BS, et al. Early onset of TNFα-driven arthritis, auto-inflammation, and progressive loss of vision in a patient with ALPK1 mutation[J]. J Clin Immunol, 2022, 42: 880-884. doi: 10.1007/s10875-022-01214-8
 下载:
下载:
计量
- 文章访问数: 581
- HTML全文浏览量: 109
- PDF下载量: 85


