 作者投稿
作者投稿 专家审稿
专家审稿 编辑办公
编辑办公 主编办公
主编办公
Clinical Characteristics and Treatment of Blau Syndrome in Chinese Children-a National Multicenter Study
-
摘要:目的
研究中国儿童Blau综合征的人口学特征、临床特点、基因型和表型的相关性及治疗情况,使该病能早期诊断并及时治疗。
方法回顾性分析全国11家中心2006年5月至2022年4月住院的中国儿童Blau综合征患者72例,收集其年龄、性别、家族史等一般信息及临床资料、化验检查及治疗用药情况。
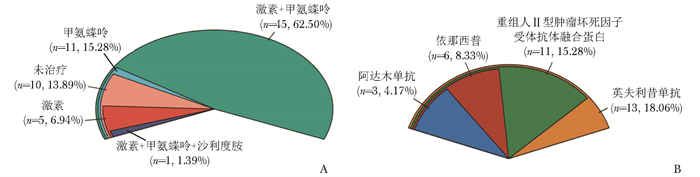
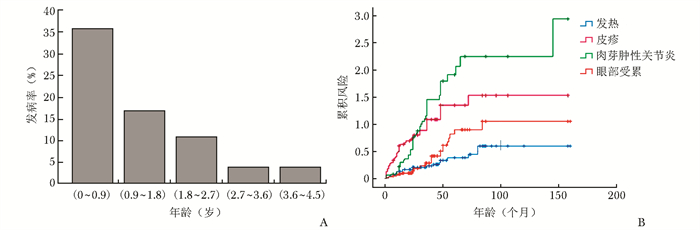
结果Blau综合征患者南北方分布较为均匀,无明显的地域倾向性。平均发病年龄(14.30±12.81)月,确诊年龄(55.18±36.22)月。35%的Blau综合征患者发病年龄在1岁之前,全部患者发病均在5岁之前。87.50%(63/72)患者出现肉芽肿性关节炎,65.28%(47/72) 患者出现皮疹,36.11%(26/72)患者出现眼部受累,27.78% (20/72)患者出现发热,15.28%(11/72)患者出现肺部受累。Blau综合征出现肉芽肿性关节炎表现的风险最大,其次为皮疹、眼部受累、发热。病程前25个月,出现皮疹的风险最大。病程25~84个月,发生关节炎的风险最大。Blau综合征主要的基因突变类型为p.R334Q和p.R334W,携带p.R334Q突变的患者有相对较高的发热[35.71%(5/14)vs. 14.29%(1/7),P=0.43]和眼部受累[42.86%(6/14)vs. 28.57% (2/7),P=0.51]发生率。在p.R334W突变的患者中,有相对较高的皮疹发生率[85.71% (6/7)vs. 64.29% (9/14),P=0.59]。45例(62.50%)患者应用糖皮质激素和甲氨蝶呤的联合治疗。22例患者在糖皮质激素和甲氨蝶呤治疗的基础上加用TNF-α拮抗剂。
结论中国儿童Blau综合征不同临床表现出现的风险由高至低依次为关节炎、皮疹、眼部受累、发热。主要治疗药物为糖皮质激素和甲氨蝶呤,可根据情况加用生物制剂。
Abstract:ObjectiveTo study the demographic and clinical characteristics, correlation of genotype and phenotype and treatment of Blau syndrome to facilitate early diagnosis and timely treatment of Blau syndrome.
MethodsSeventy-two patients with Blau syndrome from 11 centers from May 2006 to April 2022 were retrospectively analyzed, and their general information, clinical data, laboratory examination and treatment medication were collected.
ResultsThe distribution of patients with Blau syndrome was uniform in geographical north and south of China, and there was no obvious gender bias. The mean age of onset was (14.30±12.81) months, and the age of diagnosis was (55.18±36.22) months. 35% of patients with Blau syndrome happened before 1 year old, and all patients developed before 5 years old. 87.50% (63/72) had granulomatous arthritis, 65.28% (47/72) had rash, 36.11% (26/72) had ocular involvement, 27.78% (20/72) had fever, and 15.28% (11/72) had pulmonary involvement. Arthritic manifestations of Blau syndrome were most at risk, followed by rash, ocular involvement, and fever.The first 25 months of the disease, the risk of developing a rash was the greatest. The risk of developing arthritis was the greatest between 25 months and 84 months. The main mutations were p.R334Q and p.R334W, and patients with p.R334Q mutation had relatively high incidence of fever (35.71%[5/14] vs. 14.29%[1/7], P=0.43) and ocular involvement (42.86%[6/14]vs. 28.57%[2/7], P=0.51). There was a relatively high incidence of rash (85.71%[6/7] vs. 64.29%[9/14], P=0.59) in patients with the p.R334W mutation. Forty-five patients(62.50%)were treated with a combina-tion of glucocorticoid and methotrexate. Twenty-two patients were treated with tumor necrosis factor antagonist in addition to glucocorticoid and methotrexate.
ConclusionsThe risk of different clinical manifestations of Blau syndrome from high to low was arthritis, followed by rash, ocular involvement and fever. The main treatment was glucocorticoid combined with methotrexate, to which biological agents could be added.
-
Keywords:
- Blau syndrome /
- demographic characteristics /
- clinical manifestations /
- treatment
-
1. 临床资料
患儿男性,11岁10个月,主因“尿中泡沫增多2年,半月前抽搐1次”入院。2019年11月无意中发现尿中泡沫增多,无其他明显伴随症状,未予重视及处理。2021年10月患儿行走时无明显诱因出现抽搐发作,就诊于外院查生化提示:尿素45.3 mmol/L,肌酐1501 μmol/L,总钙1.47 mmol/L,血磷2.69 mmol/L,血常规示血色素64 g/L,肾脏超声示双肾缩小(右肾5.2 cm×2.5 cm,左肾5.0 cm×3.2 cm),考虑慢性肾脏病Ⅴ期,行血液净化治疗6 d后为求进一步治疗转诊至笔者医院。个人史:G1P1,孕期因宫口不开行剖宫产,无羊水及胎盘异常。听力正常,智力、运动发育正常。5岁时入学体检发现视力“发育落后”,眼科专科检查提示视盘发育不良,后予配镜400~500度,矫正视力不足1.0。家族史:父母体健,否认家族遗传性疾病病史。体格检查:体温36.6 ℃,心率85次/min,呼吸23次/min,血压109/56 mm Hg,身高145.0 cm,体重34.0 kg。神清,面色苍白,皮肤粗糙,无皮疹,无明显浮肿,听力粗测正常,心肺听诊未及明显异常。肝脾不大,移动性浊音阴性,神经系统查体未见异常。实验室检查:尿常规:尿比重1.005,蛋白(2+),潜血(2+),镜检红细胞3~5个/HPF。尿糖(+)。24 h尿蛋白定量:1095 mg/24 h。肾脏损伤标志物:尿IgG 26.7 mg/L(0~8), 尿微量白蛋白228 mg/L(0~19), 尿转铁蛋白9.91 mg/L(≤2.0),尿α1~微球蛋白207 mg/L(≤12.5),尿β2微球蛋白68 413 μg/L(≤159.7)。甲状旁腺素1153.7 pg/mL。自身抗体均阴性。铜蓝蛋白295 mg/L,补体C3、C4及凝血功能正常,泌尿系超声:左肾4.8 cm× 2.0 cm,右肾4.8 cm×2.2 cm,双肾实质弥漫不均匀增强,皮髓质分界不清,双肾散在小囊肿,大者直径为0.6 cm,双侧肾盂肾盏无扩张,双侧未见扩张的输尿管。生化:血白蛋白40.1 g/L,尿素37.7 mmol/L,肌酐825.9 μmol/L,血糖5.36 mmol/L。眼科会诊(图 1):视乳头呈粉红色,视盘面积增大约为正常视乳头面积5倍,视乳头周围有巩膜、脉络膜,视网膜萎缩环,血管走形平直,自视乳头凹陷处呈放射状向外发散。
获得患儿及其父母知情同意后,分别采集患儿及其父母外周血2 mL,进行全外显子基因测序并Sanger验证(图 2):染色体核型XY,PAX2发现有一个杂合突变c.862-1G>A,导致氨基酸发生剪接突变,NM003987。经家系验证分析,先证者父母该位点均无变异,此变异为自发突变。经美国医学遗传学和基因组学会评级指南分析,该变异为零效变异,可能导致基因功能丧失,该突变正常人群数据库中的频率为-,为低频变异。SIFT、PolyPhen_2、Mutation Taster、GERP+预测结果均为有害。
![]() 图 2 患儿及其父母基因位点变异Sanger测序图突变位点用红色箭头表示Figure 2. Sanger sequencing map of the locus variation of the patient and parents
图 2 患儿及其父母基因位点变异Sanger测序图突变位点用红色箭头表示Figure 2. Sanger sequencing map of the locus variation of the patient and parents治疗及病情进展:予低钾、低磷饮食,司维拉姆、阿法骨化醇调节钙磷代谢,左乙拉西坦预防抽搐发作。促红细胞生成素皮下注射改善贫血。经家长同意后,选择自动化腹膜透析治疗。出院前复查尿素22.35 mmol/L,肌酐759.4 μmol/L,白蛋白42.2 g/L,甲状旁腺激素648.6 pg/mL。血红蛋白93 g/L。出院后定期随诊,并完善腹膜平衡试验提示2 h肌酐低转运、2 h尿素高平均、2 h葡萄糖低转运。患儿出院后未再出现抽搐发作,血压正常,尿量900~1100 mL/d, 出院后3个月于外院行同种异体肾移植手术顺利,恢复良好。
2. 讨论
肾-视神经盘缺损综合征(renal coloboma syn-drome, RCS)也被称为视神经肾综合征(papillorenal syndrome),用来描述一种由视神经发育不良和肾脏畸形组成的疾病,遗传模式为常染色体显性遗传。1988年,Weaver等对患有视神经缺损伴终末期肾病的家系进行了首次描述,在该家系发现了常染色体PAX2基因的显性突变,证实了PAX2与RCS的相关性[1]。截至目前,全世界范围共报道250~280例患者,发病罕见。从出生至79岁之间皆可发病,绝大多数发病于儿童期[2]。92%的RCS患者存在肾脏疾病,77%存在眼部病变,最常见的肾脏表现是肾脏发育不良(65%),通常为双侧[3]。其他肾脏异常包括肾小管功能异常、囊性肾脏疾病、膀胱输尿管反流及其他先天性肾脏和尿路畸形,最终进展至终末期肾病,需要肾脏替代治疗。眼部异常包括视神经发育不良和缺损等,约占患者的72%。
PAX2基因活性与疾病表现相关,PAX2表达不足导致肾脏发育不全和输尿管畸形。迄今为止已经发现90种不同的PAX2突变和4个片段微缺失致病变异[4]。从分子角度来看,后肾间充质中表达的PAX2等转录因子汇聚于配体胶质细胞来源的神经营养因子,激活输尿管芽中酪氨酸激酶的表达,诱导其生长。从输尿管芽发出的信号启动了间充质细胞的聚集,并随后转化为肾单位的肾小管上皮细胞。随着发育,PAX2表达下调,在成熟的集合管中很少有表达,在肾小球中始终不表达[3]。故PAX2异常多影响肾脏尤其是肾小管发育,导致肾小管、间质及输尿管结构和功能异常而肾小球基本不受影响。本例患儿表现为肾小管功能异常,但病程中始终无浮肿、大量蛋白尿、肉眼血尿等肾小球损害表现,符合PAX2基因临床特点。但需要说明的是,患儿双肾萎缩存在慢性肾脏病因素,但是否同时合并双肾发育不良,本例缺乏临床资料的支持。
不少罕见病存在肾脏及眼部的损害,由于症状隐匿及表现时间不同,容易出现遗漏诊治的情况。本例患儿在6岁时眼科检查发现视力异常及视盘发育异常,在11岁时出现肾脏症状时已处于尿毒症期。又如肾小管实质性肾病-视网膜变性综合征(Senior-Loken综合征):表现为生后逐渐进展的视网膜退行性病变,肾脏早期多为烦渴、多尿、多饮,轻微尿检异常,在13~15岁出现肾功能衰竭;还有如眼-脑-肾综合征(lowe综合征):眼部以先天性白内障及先天性青光眼常见,随着病情进展,逐步出现Fanconi综合征及不同程度的肾功能减退。此外,胱氨酸贮积症:患儿多在10岁前进展至终末期肾病,虽然1岁时眼裂隙灯检查可见角膜结晶沉积,明显的视力缺损多发生在10~20岁,尤其是15岁以后[5]。本例提示当患者存在眼部发育异常时需及时监测肾功能,或当患者存在肾脏发育不良时进行眼部检查。对于早期明确诊断及指导治疗具有重要意义。
随着遗传学技术发展,越来越多的泌尿系统遗传性罕见疾病被发现和诊断,其中多数疾病被认为是慢性肾功能衰竭的重要原因[6]。因此,对于罕见肾脏疾病的认识更加需要重视。不能仅停留在诊断慢性肾脏病的基础上,积极完善基因检查寻找病因,对于明确诊断、监测肾外症状及改善患儿预后具有重要作用。如本例患儿在诊断慢性肾衰竭后早期完善了基因检查,发现PAX2基因的致病突变,明确了肾-视神经盘缺损综合征的诊断,解释了患儿的眼部及肾脏病变,对于开展眼部护理及肾替代治疗有重要的指导意义。
目前报道部分RCS患者经过一次或两次肾移植后状态良好,个别患者死于移植后相关并发症[1]。本例患儿于诊断后6个月行肾移植术,目前术后恢复良好。对于遗传因素所致肾脏疾病,移植后多不存在原发病复发的风险,移植肾存活时间较长[7]。但需要注意的是,部分累及肾脏的罕见疾病,随着肾移植后寿命延长,肾外症状逐渐发展加重,并且成为影响患儿远期预后及生活质量的重要因素。比如胱氨酸贮积症,随着年龄的增长,逐渐出现糖尿病、视力减退、甲状腺功能减退等问题,需要积极应用半胱胺滴眼液降低结晶沉积,监测并调节血糖、甲状腺功能等。
综上所述,肾-视神经盘缺损综合征为罕见的常染色体显性遗传病,以肾和眼的发育及功能异常为主要临床特点,视力异常多为早期表现,肾脏症状可能起病隐匿,缓慢进展。对于临床上发现不明原因的肾脏、眼部发育及功能异常的患儿要积极完善基因检查,有助于疾病的确诊、监测和治疗。
作者贡献:李彩凤负责研究设计;张俊梅、赵晓珍负责数据分析、论文撰写;唐雪梅、赵伊楠、李丽、高凤乔、史昕炜、金燕樑、张宇、曹兰芳、尹薇、肖继红、邝伟英、邓江红、王江、檀晓华、李超、李士朋、薛海燕、刘翠华、刘小惠、赵冬梅、陈雨青、郑雯洁负责临床资料收集。利益冲突:所有作者均声明不存在利益冲突。 -
![]()
图 1 Blau综合征患者的发病率
A. Blau综合征总发病率;B. Blau综合征不同临床表现的风险预测
Figure 1. Incidence in patients with Blau syndrome
![]()
图 2 72例Blau综合征患者的治疗现状
A.传统治疗的现状; B.生物制剂应用的现状
Figure 2. Treatment status of 72 patients with Blau syndrome
表 1 72例儿童Blau综合征患者的人口学和临床特征
Table 1 Demographic and clinical characteristics of 72 children with Blau syndrome
指标 例数 百分比(%) 籍贯:北方 39 54.17 家族史 17 23.61 性别:女 32 44.44 发病年龄(x±s,个月) 14.30±12.81 - 确诊年龄(x±s,个月) 55.18±36.22 - 发热 20 27.78 皮疹 47 65.28 肉芽肿性关节炎 63 87.50 眼部受累 26 36.11 高血压 1 1.39 骨软骨瘤 2 2.78 听力损害 4 5.56 肾脏受累 1 1.39 血管炎 24 33.33 心脏受累 4 5.56 肺部受累 11 15.28 中枢神经系统受累 2 2.78 中国北方:中国南北方划分以秦岭——淮河为界,此线以北,定义为北方  下载: 导出CSV
下载: 导出CSV
-
[1] 李彩凤. 儿童Blau综合征研究进展[J]. 中国实用儿科杂志, 2018, 33: 26-29. https://www.cnki.com.cn/Article/CJFDTOTAL-ZSEK201801010.htm [2] Rosé CD, Doyle TM, McIlvain-Simpson G, et al. Blau syndrome mutation of CARD15/NOD2 in sporadic early onset granulomatous arthritis[J]. J Rheumatol, 2005, 32: 373-375.
[3] Rosé CD, Wouters CH, Meiorin S, et al. Pediatric granulomatous arthritis: an international registry[J]. Arthritis Rheum, 2006, 54: 3337-3344. doi: 10.1002/art.22122
[4] Aróstegui JI, Arnal C, Merino R, et al. NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort[J]. Arthritis Rheum, 2007, 56: 3805-3813. doi: 10.1002/art.22966
[5] Sfriso P, Caso F, Tognon S, et al. Blau syndrome, clinical and genetic aspects[J]. Autoimmun Rev, 2012, 12: 44-51. doi: 10.1016/j.autrev.2012.07.028
[6] Punzi L, Gava A, Galozzi P, et al. Miscellaneous non-inflammatory musculoskeletal conditions. Blau syndrome[J]. Best Pract Res Clin Rheumatol, 2011, 25: 703-714.
[7] Clark SK. Sarcoidosis in children[J]. Pediatr Dermatol, 1987, 4: 291-299. doi: 10.1111/j.1525-1470.1987.tb00796.x
[8] Raphael SA, Blau EB, Zhang WH, et al. Analysis of a large kindred with Blau syndrome for HLA, autoimmunity, and sarcoidosis[J]. Am J Dis Child, 1993, 147: 842-848.
[9] Manouvrier-Hanu S, Puech B, Piette F, et al. Blau syndrome of granulomatous arthritis, iritis, and skin rash: a new family and review of the literature[J]. Am J Med Genet, 1998, 76: 217-221. doi: 10.1002/(SICI)1096-8628(19980319)76:3<217::AID-AJMG4>3.0.CO;2-N
[10] Ikeda K, Seto Y, Narita A, et al. Ultrasound assessment of synovial pathologic features in rheumatoid arthritis using comprehensive multiplane images of the second metacarpophalangeal joint: identification of the components that are reliable and influential on the global assessment of the whole joint[J]. Arthritis Rheumatol, 2014, 66: 523-532.
[11] Okafuji I, Nishikomori R, Kanazawa N, et al. Role of the NOD2 genotype in the clinical phenotype of Blau syndrome and early-onset sarcoidosis[J]. Arthritis Rheum, 2009, 60: 242-250. doi: 10.1002/art.24134
[12] Kurokawa T, Kikuchi T, Ohta K, et al. Ocular manifestations in Blau syndrome associated with a CARD15/Nod2 mutation[J]. Ophthalmology, 2003, 110: 2040-2044. doi: 10.1016/S0161-6420(03)00717-6
[13] Miceli-Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome[J]. Nat Genet, 2001, 29: 19-20. doi: 10.1038/ng720
[14] Ogura Y, Inohara N, Benito A, et al. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB[J]. J Biol Chem, 2001, 276: 4812-4818. doi: 10.1074/jbc.M008072200
[15] Girardin SE, Boneca IG, Viala J, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP)detection[J]. J Biol Chem, 2003, 278: 8869-8872. doi: 10.1074/jbc.C200651200
[16] Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease[J]. J Biol Chem, 2003, 278: 5509-5512.
[17] Kanazawa N, Okafuji I, Kambe N, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome[J]. Blood, 2005, 105: 1195-1197.
[18] Chamaillard M, Philpott D, Girardin SE, et al. Gene-environment interaction modulated by allelic heterogeneity in inflammatory diseases[J]. Proc Natl Acad Sci USA, 2003, 100: 3455-3460. doi: 10.1073/pnas.0530276100
[19] 李彩凤, 张俊梅, 王江, 等. 中国儿童肉芽肿性关节炎26例临床特点及治疗随访分析[J]. 中华风湿病学杂志, 2014, 18: 593-596, 652. [20] Li C, Zhang J, Li S, et al. Gene mutations and clinical phenotypes in Chinese children with Blau syndrome[J]. Sci China Life Sci, 2017, 60: 758-762.
-
期刊类型引用(1)
1. 马雪晴,何永华,杨静,徐荣荣,杨思莹,梁文沛,周建华,袁惠卿,仇丽茹. 中国儿童 PAX2基因突变的临床表型和基因型分析. 中华肾脏病杂志. 2024(01): 24-35 .  百度学术
百度学术
其他类型引用(0)


计量
- 文章访问数: 684
- HTML全文浏览量: 281
- PDF下载量: 81
- 被引次数: 1


