 作者投稿
作者投稿 专家审稿
专家审稿 编辑办公
编辑办公 主编办公
主编办公
-
摘要:
Erdheim-Chester病(Erdheim-Chester disease,ECD)是一种罕见的非朗格汉斯细胞组织细胞增生症,其主要特征是富含脂质的泡沫细胞在骨骼及多器官系统浸润,临床表现复杂多样,容易误诊。本文报道一例因下丘脑功能紊乱就诊,中枢神经系统、胰腺及下肢骨受累,经临床、影像、病理综合诊断为ECD的患者,干扰素α治疗后临床症状及影像表现均明显好转。
-
关键词:
- Erdheim-Chester病 /
- 组织细胞增多症 /
- 磁共振成像
Abstract:Erdheim-Chester disease (ECD) is a rare non-Langerhans cell histiocytosis characterized by the infiltration of bone and multiple organs by foamy lipid-laden histiocytes. ECD is easy to be misdiagnosed due to its complicated clinical manifestations. We report a patient who visited the hospital due to hypothalamic dysfunction, with central nervous system, pancreas, and lower limb bones involvements. Together with the evidence of clinical manifestations, imaging and pathology, this patient was diagnosed with ECD. After treatment with interferon-α, both the clinical symptoms and imaging manifestations of this patient were significantly improved.
-
Keywords:
- Erdheim-Chester disease /
- histiocytosis /
- magnetic resonance imaging
-
1. 病例简介
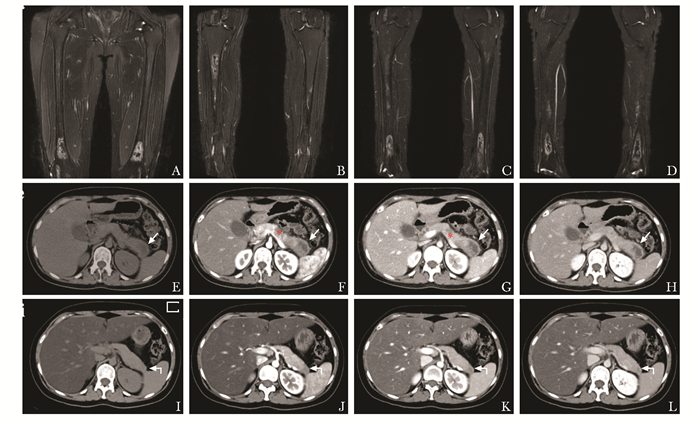
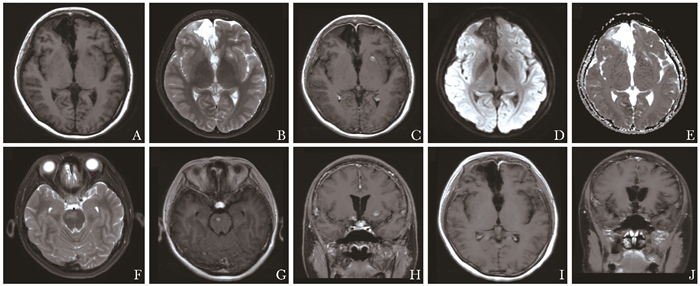
患者女,29岁,因月经不调6年、多饮多尿2年入院。6年前,患者因月经不调就诊于外院,予激素、中药治疗(具体不详)后,症状有所好转。2年前,患者出现烦渴、尿量明显增多,外院头部MRI检查提示“鞍区占位”。入我院后,行头部增强MRI检查,发现下丘脑、鞍上、左壳核及桥脑多发结节影,呈稍长T1长T2信号,DWI信号不高,增强后病变呈Erdheim-Chester病一例明显均匀强化,脑内病灶伴瘤周水肿(图 1A~H)。双下肢MRI平扫示双侧股骨及胫骨多发地图样异常信号,考虑骨梗死(图 2A~D)。腹部增强CT显示胰体尾多发片状低密度影,增强后延迟强化,较正常胰腺实质强化减低,脾静脉局部受累(图 2E~H)。血生化检查:睾酮<0.1 μg/L(0.1~ 0.75 μg/L)卵泡刺激素、黄体生成素、雌二醇、孕酮及泌乳素正常;促甲状腺激素<0.008 mU/L(0.38~ 4.34 mU/L), 游离T3、游离T4正常;皮质醇<5 μg/L(40~223 μg/L),促肾上腺皮质激素及生长激素正常。行鞍区肿物切除术,术后病理提示:组织细胞增生症,免疫组化S100(+)、Vimentin(+)、MGMT (+)、Ki67(约5%), CD1a、Langerin、CD34、ALK、EMA、CgA、GFAP、P53、Syn、SMA、PLAP均为阴性,BRAF V600E突变阴性。行超声内镜检查,显示胰体尾部多发低回声实性病变,边界不清,形态不规则,胰管规整。在超声内镜下行胰腺细针穿刺,细胞学涂片提示:未见恶性肿瘤细胞。结合患者临床症状、影像学表现、病理结果及免疫组化,诊断为Erdheim-Chester病。予重组人干扰素α-2a皮下注射治疗9个月后复查,患者症状减轻,复查头增强MRI及腹部增强CT显示病灶均较前明显减小(图 1I~J、图 2I~L)。
![]() 图 1 头MRI结果左基底节区(A~E)、桥脑(F~G)、鞍区及下丘脑(H)多发占位,病灶呈稍长T1(A)、长T2信号(B, F),脑内病灶伴瘤周水肿,DWI信号不高(D),ADC未见减低(E),增强后病变呈明显均匀强化(C, G~H); 鞍区肿物切除术后,右前额叶为术后改变,局部软化灶(A~E, I);干扰素治疗9个月后,T1增强序列示基底节区病灶消失(I),鞍区及下丘脑病灶明显减小(J)
图 1 头MRI结果左基底节区(A~E)、桥脑(F~G)、鞍区及下丘脑(H)多发占位,病灶呈稍长T1(A)、长T2信号(B, F),脑内病灶伴瘤周水肿,DWI信号不高(D),ADC未见减低(E),增强后病变呈明显均匀强化(C, G~H); 鞍区肿物切除术后,右前额叶为术后改变,局部软化灶(A~E, I);干扰素治疗9个月后,T1增强序列示基底节区病灶消失(I),鞍区及下丘脑病灶明显减小(J)![]() 图 2 双下肢MRI和腹部增强CT结果MRI结果显示双侧股骨(A)、胫骨(B~D)骨干及干骺端对称性骨梗死;腹部增强CT示胰体尾部多发低密度影(E,箭头),增强扫描示动脉期(F)、静脉期(G)至延迟期(H)逐渐强化,各期均低于周围正常胰腺实质,局部脾静脉狭窄(*);治疗9个月后复查,病灶明显减小(I~L,弯箭头)
图 2 双下肢MRI和腹部增强CT结果MRI结果显示双侧股骨(A)、胫骨(B~D)骨干及干骺端对称性骨梗死;腹部增强CT示胰体尾部多发低密度影(E,箭头),增强扫描示动脉期(F)、静脉期(G)至延迟期(H)逐渐强化,各期均低于周围正常胰腺实质,局部脾静脉狭窄(*);治疗9个月后复查,病灶明显减小(I~L,弯箭头)2. 讨论
William Chester和Jakob Erdheim首次描述了一种罕见的非朗格汉斯细胞组织细胞增生症[1],该疾病在1972年被首次命名为ECD,并在2016年被世界卫生组织(World Health Organization,WHO)列为组织细胞疾病中与朗格汉斯组织细胞增生症(Langerhans cell histocytosis,LCH)并列的,非朗格汉斯细胞组织细胞病中单独的一类。到目前为止,全球共报道约1500例[2]。本病好发于中老年人(中位年龄55岁),男性较多(男女比例约为3∶1),儿童少见[2]。本病常为多系统受累,临床表现异质性较高。当该病首发症状为骨痛、神经系统症状及尿崩症时,容易被误诊[3]。
本文中所报道的这例病例累及下丘脑-垂体轴、脑实质、下肢骨及胰腺,但首发症状为月经失调、尿崩症,故首先要与LCH鉴别。两者累及下丘脑-垂体轴时都可表现为垂体柄增粗、鞍区及下丘脑占位和垂体后叶短T1信号消失[3-5]。两者的鉴别点为:LCH在儿童中更常见,ECD好发于老年人;与LCH相比,ECD中枢神经系统受累合并下丘脑-垂体轴外的病灶更多见[6];颅骨受累在LCH中常表现为溶骨性骨质破坏[7],ECD则多表现为骨质增厚、硬化[8]。
ECD的中枢神经系统受累通常表现为3种形式:肿瘤样病变、血管受累和退行性变[9]。肿瘤样病变可单发或多发,累及部位广泛,硬脑膜、幕上脑实质、脉络丛、脑室旁区域、脑干、小脑均可受累。脑内的病灶常呈结节样,T1WI呈等或稍低信号,T2WI呈高信号,静脉注射对比剂增强后呈明显强化[8]。本例患者为青年女性,除下丘脑-垂体轴受累外,基底节区及脑干可见多发结节,直径6~9 mm,信号特点与既往文献报道一致[8]。另外,本例病例脑实质内病灶可见瘤周水肿,先前我们的队列也报道过1例灶周明显水肿[10],这一现象在既往文献中报道较少,易误诊为转移瘤。此外,ECD的肿瘤样病变不伴扩散受限,DWI信号不高。ECD血管受累可表现为血管周围肿瘤浸润,引起血管管腔狭窄及卒中。退行性变常表现为脑干或小脑T2WI/FLAIR高信号,增强后无强化,以及大脑皮层或小脑萎缩[9]。此外,在ECD患者的头部MRI检查中,有时还能观察到鼻窦及眼眶受累、颅骨增厚[8]。本例ECD患者的中枢神经系统受累仅表现为肿瘤样病变,无血管受累、退行性变、鼻窦、眼眶受累及颅骨增厚等改变。
高达80%~95%的ECD患者有骨骼病变,但只有约38%的患者出现骨痛等临床症状[2]。ECD骨病主要表现为四肢长骨的对称性骨硬化,典型病变为膝关节周围(股骨下段及胫骨上段)骨质密度增高[11]。大部分患者的四肢骨病变双侧对称,少数患者可在双侧病变的基础上出现单侧病变,如单侧肢体长骨的骨膜炎。除四肢骨外,少部分患者还可累及颅颌面骨和副鼻窦区,但鲜有脊柱受累[11]。本例患者无骨骼相关症状,但表现为双下肢骨大致对称性分布的多发骨梗死灶。X线、CT、MRI、骨扫描均可显示病灶,骨扫描可见病变部位对示踪剂摄取增加,且对称性分布于膝关节周围,此表现具有特征性[11]。当影像学检查发现长骨病变时,对本病有重要的提示意义。
此外,本例患者可见胰腺受累,这也是本例ECD诊断过程中的难点。增强CT可见胰体尾片状低密度影,延迟低强化,并累及邻近的脾静脉。复习既往文献,尚未见ECD累及胰腺的报道,故该患者的胰腺病变首先要与胰腺癌鉴别。本例行超声内镜检查,显示胰体尾部多发低回声实性病变,边界不清,形态不规则,胰管规整。并在超声内镜引导下行扇形穿刺胰腺多个病灶,细胞学涂片未见恶性肿瘤细胞。结合患者的临床症状、对称性下肢骨受累、中枢神经系统病变及病理,从“一元论”角度考虑该患者胰腺病变为ECD累及可能。使用重组人干扰素α-2a治疗9个月时复查,患者临床症状改善,影像学检查显示颅内病灶及胰腺病灶较前明显减少、减小,表明干扰素治疗有效。这也进一步证实了胰腺病灶为ECD所致可能性大。
除神经系统、骨及胰腺受累外,ECD还可累及心血管系统,常见表现包括心包积液或心包填塞、右房室假肿瘤性心肌浸润、主动脉鞘、肾动脉受累致高血压等;累及呼吸系统,胸部CT表现为纵隔浸润、胸膜增厚或积液、小叶间隔增厚、磨玻璃影或小叶中心结节;腹膜后受累常表现为“毛发肾”,并可向肾盂或输尿管延伸,导致肾积水和肾功能衰竭;皮肤受累多表现为黄瘤、皮下结节或环状肉芽肿样变[11]。本例ECD患者未见上述器官系统受累。
本病例提示,青年患者以尿崩症起病时,应考虑到ECD的可能诊断,积极寻找神经系统及其他器官系统受累的证据,对称性下肢骨受累对本病诊断有提示意义。此外,ECD亦可累及胰腺,CT表现为低密度灶伴延迟低强化,可伴邻近血管受累,结合全身多系统影像学表现及病理学检查,可与胰腺癌鉴别诊断。
综上所述,ECD是一种罕见的多系统受累的组织细胞增生性疾病,临床症状及影像学表现多样, 需仔细询问病史,全面了解患者的临床症状,并进行有针对性的影像学检查确定病变和活检部位,识别典型和不典型的影像学表现,进行临床-影像-病理综合诊断。
作者贡献:范晓媛:调查、形式分析、可视化、撰写初稿;刘婷:数据管理、形式分析;有慧:形式分析、方法;孙健:形式分析、方法;曹欣欣:资源、项目管理、监督;冯逢:概念化、资源、初稿审阅及修订;王凤丹:概念化、资助、监督、初稿审阅及修订。利益冲突:所有作者均声明不存在利益冲突 -
![]()
图 1 头MRI结果
左基底节区(A~E)、桥脑(F~G)、鞍区及下丘脑(H)多发占位,病灶呈稍长T1(A)、长T2信号(B, F),脑内病灶伴瘤周水肿,DWI信号不高(D),ADC未见减低(E),增强后病变呈明显均匀强化(C, G~H); 鞍区肿物切除术后,右前额叶为术后改变,局部软化灶(A~E, I);干扰素治疗9个月后,T1增强序列示基底节区病灶消失(I),鞍区及下丘脑病灶明显减小(J)
-
[1] Chester W. Über lipoidgranulomatose [J]. Virchows Arch pathol Anat, 1930, 279: 561-602. doi: 10.1007/BF01942684
[2] Haroche J, Cohen-Aubart F, Amoura Z. Erdheim-Chester disease [J]. Blood, 2020, 135: 1311-1318. doi: 10.1182/blood.2019002766
[3] Shekhar S, Irizarry-Caro JA, Sinaii N, et al. Pituitary imaging abnormalities and related endocrine disorders in Erdheim-Chester disease [J]. Cancers, 2021, 13: 4126. doi: 10.3390/cancers13164126
[4] Courtillot C, LaugierRobiolle S, Cohen Aubart F, et al. Endocrine manifestations in a monocentric cohort of 64 patients with Erdheim-Chester disease [J]. J Clin Endocrinol Metab, 2016, 101: 305-313. doi: 10.1210/jc.2015-3357
[5] Prayer D, Grois N, Prosch H, et al. MR imaging presentation of intracranial disease associated with langerhanscell histiocytosis [J]. AJNR Am J Neuroradiol 2004, 25: 880-891.
[6] Cohen Aubart F, Idbaih A, Emile JF, et al. Histiocytosis and the nervous system: from diagnosis to targeted therapies [J]. Neuro Oncol, 2021, 23: 1433-1446. doi: 10.1093/neuonc/noab107
[7] Grois N, Fahrner B, Arceci RJ, et al. Central nervous system disease in langerhanscell histiocytosis [J]. J Pediatr, 2010, 156: 873-881. doi: 10.1016/j.jpeds.2010.03.001
[8] Drier A, Haroche J, Savatovsky J, et al. Cerebral, facial, and orbital involvement in Erdheim-Chester disease: CT and MR imaging findings [J]. Radiology, 2010, 255: 586-594. doi: 10.1148/radiol.10090320
[9] Cohen Aubart F, Idbaih A, Galanaud D, et al. Central nervous system involvement in Erdheim-Chester disease: An observational cohort study [J]. Neurology, 2020, 95: e2746-e2754. doi: 10.1212/WNL.0000000000010748
[10] Wang F, Cao X, Niu N, et al. Multisystemic imaging findings in Chinese patients with Erdheim-Chester disease [J]. AJR Am J Roentgenol, 2019, 213: 1179-1186. doi: 10.2214/AJR.19.21523
[11] Goyal G, Heaney ML, Collin M, et al. Erdheim-Chester disease: consensus recommendations for evaluation, diagnosis, and treatment in the molecular era [J]. Blood, 2020, 135: 1929-1945. doi: 10.1182/blood.2019003507
-
期刊类型引用(1)
1. 陈其春,王龙胜,赵红,吴旱秋. Erdheim-Chester病影像诊断——2022年读片窗(12). 安徽医学. 2022(12): 1487-1489 .  百度学术
百度学术
其他类型引用(0)
 下载:
下载:
计量
- 文章访问数: 789
- HTML全文浏览量: 291
- PDF下载量: 128
- 被引次数: 1


