 作者投稿
作者投稿 专家审稿
专家审稿 编辑办公
编辑办公 主编办公
主编办公
To Identify High-Risk Adolescent and Adult Spinal Muscular Atrophy Populations: Exploration of Methods and Perspectives
-
摘要:
脊髓性肌萎缩症(SMA)是一种罕见的遗传性神经肌肉病,患者临床异质性较大。根据病情轻重和进展速度共分为5型。近年来随着多学科综合管理的推广和疾病修正治疗药物的应用,SMA患者的预后已明显改善,更多患者进入青少年及成人期。不同类型患者在青少年和成人期的状态各不相同,也使得这一年龄段患者表现更加复杂多样,识别与诊断更为困难。中国幅员辽阔、人口众多,不同地区医疗水平不均衡,进一步加剧了青少年及成人SMA患者的诊疗难题,误诊或诊断延迟仍是许多患者未解决的首要问题。患者就诊时首诊科室分布广泛,加强非神经肌肉病专科医生对青少年及成人SMA高危人群的识别具有重要意义。本文尝试探讨一种简单清晰、可操作性强的青少年成人SMA高危人群“画像”式识别方法,以期帮助可能首诊SMA患者的非神经肌肉病专科医生做到早期识别、早期诊断,使患者尽早获得规范治疗,从而进一步提高患者的临床获益,改善患者及其家庭的生活质量。
Abstract:Spinal muscular atrophy(SMA)is a rare genetic neuromuscular disease characterized by significant clinical heterogeneity among patients. According to the severity and progression rate of the condition, the disease is classified into five types. In recent years, because of the promotion of multidisciplinary management and the application of disease-modifying therapies, the prognosis of SMA patients has significantly improved, resulting in more patients entering into the stage of adolescence and adulthood. The varying conditions of different types of patients in the adolescence and adulthood make the manifestations more complex and diverse, leading to the difficulty in identification and diagnosis. Because of the vast territory and large population in China, coupled with uneven health care development among different regions of the country, the diagnosis and treatment for adolescent and adult SMA patients are very challenging. Misdiagnosis or delayed diagnosis remains a primary unresolved issue for many patients. The fact that patients have to visit various departments in their initial consultation highlights the importance of enhancing the recognition of high-risk adolescent and adult SMA populations among the non-neuromuscular specialists. This article attempts to explore a simple, clear, and highly operational "portrait" way of identifying the high-risk adolescent and adult SMA patients in the population, aiming at assisting the non-neuromuscular specialists to diagnose SMA patients in a way of early recognition and diagnosis and to ensure patients receiving standardized treatment as early as possible. The ultimate goal is for the higher clinical gain and a better life for patients and their families.
-
脊髓性肌萎缩症(spinal muscular atrophy,SMA)是一种罕见的常染色体隐性遗传病,由5号染色体长臂上的运动神经元存活(survival motor neuron,SMN)基因1(SMN1)纯合缺失或复合杂合突变导致脊髓前角运动神经元和低位脑干运动神经核变性,进而出现肌肉进行性无力、萎缩[1],也被称为5q-SMA。SMA的起病年龄差异大,婴幼儿、儿童、青少年和成人阶段均可发病。该病的全球发病率在活产婴儿中为(7.8~10)/10万例[1],是遗传病所致的2岁以内婴儿死亡的首要原因。而随着疾病修正治疗药物(如利司扑兰、诺西那生)和基因治疗药物(如Onasemno-gene abeparvovec)等新兴治疗的发展,越来越多的SMA患者能够存活至青少年及成年期。据报道,目前全球青少年和成人SMA患者已分别约占所有SMA患者的21%和26%[2]。尽管SMA进展缓慢,但该病仍会给青少年和成人SMA患者带来明显的影响,包括活动受限、脊柱侧凸、关节挛缩、呼吸衰竭和预期寿命缩短等[3], 因此SMA患者需要终生接受治疗,面临着身体、心理、经济和社会等多方面负担。
然而,国内外对于青少年和成人SMA患者的认识并不多,很多诊治问题尚无明确解决方案。为此,来自中国多个SMA诊疗中心的多学科专家根据现有规范诊治证据并结合临床实践经验,经充分讨论,于2023年发表了《青少年成人脊髓性肌萎缩症临床诊疗指南》(后文简称“青少年成人SMA指南”)[4],为规范化临床管理提供了重要参考。由于青少年和成人SMA患者常起病隐匿、进展缓慢,加之患者分布广泛、各地医疗资源水平不均衡等因素,临床实际工作中SMA误诊、漏诊或延迟诊断的现象仍很常见。SMA患者临床症状不典型,除在神经科或小儿神经科就诊之外,可能在多个科室就诊,包括骨科、呼吸科、康复科、中医科和普通内科等。因此,加强非神经肌肉病专科医生对青少年和成人SMA高危人群的识别非常重要,有助于SMA的早诊早治,缩短诊断流程,及时早期治疗,从而改善患者预后。
1. 青少年和成人脊髓性肌萎缩症的诊断现状
根据起病年龄及可达到的最大运动能力,SMA国际分型将该病分为0~4型并细分多种亚型[4]。0型患儿出生即有明显临床表现,且常在出生后几周内死亡[5]。1型SMA患儿出生后6个月内起病,最大运动能力(运动里程碑)仅为部分抬头能力,不能独坐;2型SMA发病年龄为6~18月龄,运动里程碑为可独坐,但不能独站独走;3型SMA发病年龄为18月龄以上,患者保有或曾经获得独站独走能力;4型SMA通常为成年发病(>21岁),具备跑跳等所有运动能力[4, 6]。流行病学调查显示,绝大多数2型和3型SMA患者,甚至部分1型患者均可存活至青少年及成人期[7]。
确诊SMA需要基因诊断,国内外指南/共识均指出,应对疑似SMA的患者进行基因检测[4, 8]。而对于如何识别这些高危或疑似患者,目前形成的共识主要针对婴幼儿/儿童人群,如软婴综合征需首先考虑SMA[4]。另外,新生儿SMA筛查已在部分国家得到推广并发挥了积极作用,中国也已在部分省市试点开展。患儿在症状前期就被筛查发现,从而进入随诊治疗流程[9-10]。近年来,孕前、产前SMA基因筛查也在国内外备受关注[11-12]。因此,婴幼儿/儿童SMA患者漏诊少。与之形成鲜明对比的是青少年成人SMA患者,目前并无公认的高危人群识别工具。
青少年成人SMA患者具有起病隐匿、进展缓慢、病程长等特点,识别诊断更为困难[4]。此类患者病程长达数年甚至数十年,进展至中晚期后,常与遗传性肌肉病难以区分[13]。4型SMA也常被误诊为肢带型肌营养不良、脊髓延髓性肌萎缩症或其他运动神经元病[14]。同时,由于症状不典型,青少年及成人SMA患者常因出现步态异常、瘦弱、呼吸功能不全等问题首先在骨科、中医科、呼吸科、康复理疗科等其他非神经科就诊;而这些科室的医生对SMA认识更为不足,导致诊断延迟。即使在神经科就诊,由于神经科亚专科众多,神经肌肉病专科医师占比有限,也可能由于就诊于非神经肌肉病专科医生而未获得及时诊治。
从全球近年来报道的青少年及成人SMA误诊、诊断延迟数据来看,这一情况不容乐观。在Souza等[14]报告的一项横断面研究中,其纳入的所有20例4型SMA患者在确诊前均被误诊为其他神经系统疾病,其中60%为肢带型肌营养不良,20%为肌萎缩侧索硬化,10%为炎性肌病,10%为慢性炎症性脱髓鞘性多发性神经根神经病。大型Cure SMA调查[6]结果发现,美国患者中1型SMA患者的中位诊断延迟时间为2个月,而3型SMA的中位诊断延迟时间则长达37个月。另一项基于21项SMA诊断延迟相关研究的系统性综述结果也显示,1、2和3型SMA患者的中位诊断延迟时间(按患者人数加权后)分别为3.6、14.3和43.6个月[15];该系统性综述的亚组分析结果还发现,相比较北美患者,亚太地区和欧洲患者的确诊时间更晚[15]。在中国,2021年一项基于国家罕见病注册系统(National Rare Diseases Registry System,NRDRS)的回顾性队列研究结果显示,1、2和3型SMA患者诊断时间窗的中位数分别为3.38个月[四分位距(interquartile range,IQR):2.01~4.98]、4.08个月(IQR:2.07~8.17)和11.37个月(IQR:4.92~24.07)[16]。同时,中国的实际情况决定患者分布分散、各地医疗水平和资源不均衡,以及医务工作者对SMA疾病认识不足、诊治不及时的情况更为突出。来自中国罕见病联盟的一项调查显示,在接受调研的38 634名医务工作者中,近70%认为自己并不了解罕见病[17]。在2023年10月发布的《中国医生脊髓性肌萎缩症疾病认知与诊疗现状蓝皮书》的调研项目中,只有25%的受访医生认为自己有独立诊断SMA的能力,29%的医生需在高年资医生或多学科诊疗团队协作下才能完成SMA的最终诊断。由此可知,中国SMA患者的误诊或延迟诊断仍是普遍存在的问题。
延迟诊断带来的后果就是针对性治疗随之延迟,患者治疗获益下降,目前已公认在运动神经元仍较多存活时(如SMA患者症状前阶段)进行治疗是干预和治疗最有效的时间窗口。SUNFISH研究[18]和相关真实世界研究[19]证实,青少年成人SMA患者可从利司扑兰的治疗中获得有临床意义的运动功能和生活质量改善。NURTURE研究[20]结果也显示,73%接受了诺西那生治疗的SMA婴儿能够在健康儿童获得独走能力的预期年龄窗口内实现独立行走。SPR1NT研究[21]结果也表明,出现症状前接受Onasemnogene abeparvovec治疗的SMA儿童,达到了符合年龄的运动里程碑(包括站立和行走)。这些证据均支持早诊断早治疗是改善患者预后的关键。
为实现青少年成人SMA患者的早诊早治,加强临床医师对SMA全面认识的继续教育是一项需长久坚持的举措,但需要较长周期和持续努力。与此同时,如何能够准确、快速地识别出青少年成人SMA高危人群,并转诊到有诊治经验、能为其确诊的神经肌肉病专科医师及相应科室、医院,是减少青少年成人SMA患者诊断延迟的有效方法。
2. 如何帮助临床医师识别脊髓性肌萎缩症高危人群或疑似患者
2.1 青少年成人SMA高危人群/疑似患者的可能表现及特征
2.1.1 家族史
从SMA的致病原因来看,该病是由SMN1基因纯合缺失或复合杂合突变引起的常染色体隐性遗传性疾病,约95%的SMA患者存在SMN1纯合缺失[22]。《中国SMA患者生存现状报告白皮书》中提出,当父母均为致病基因携带者时,每次生育SMA患儿的风险为25%,且男女患病机会均等。一项基于中国227例SMA患儿家族的SMN1和SMN2基因分析结果发现,当父母的SMN2拷贝数均为1时,75%的SMA患儿为1型,25%为2型或3型;当父母的SMN2拷贝数均为3时,其所有SMA子女均为2型或3型;遗传自父母的SMN2拷贝数部分决定了SMA患儿疾病的严重程度[22]。虽然对于常染色体隐性遗传疾病,仅有少数患者家庭存在明确家族史,但基于遗传病的自然特点和对病情轻重判断的帮助,询问家族史成为评估患病可能性及疾病严重程度最简单快速的判断依据之一。
2.1.2 临床评估
青少年成人SMA指南指出,青少年及成人SMA患者虽表现为运动发育略落后,但通常可获得重要运动里程碑,随后出现进行性对称性肌无力症状,运动功能逐步减退——从下肢近端开始,以蹲起、上楼能力下降为初始表现,再向上肢近端、四肢远端及头颈部、躯干发展[4]。同时,患者常会体验到明显疲劳感,影响日常功能和生活质量[13]。相对而言,患者腿部受到的影响比手臂更早、更显著,后期可出现张口困难、吞咽力弱等[8]。2型SMA患者起初抓握物体的能力常不受影响,这与下肢无力形成鲜明对比;少数严重患者也会在早期发现胸廓变形,表现为胸部呈钟形或不对称[23]。3型SMA患者晚期可因远端肢体无力而出现高足弓等关节畸形,或因前臂肌肉无力和关节挛缩而导致手部震颤和姿势性不自主运动[13]。此外,患者查体可观察到腱反射减低或消失,病理征则为阴性[4]。
肌酸激酶(creatine kinase,CK)对维持骨骼肌能量代谢至关重要,可催化磷酸肌酸的生成,使其作为能量分子储备于骨骼肌[24]。在大多数SMA患者中,尤其是处于病情活跃进展期的患者,常可观察到CK水平呈轻中度升高(通常在正常上限3倍以内),这与失去运动神经元支配的骨骼肌细胞凋亡崩解相关[24]。SMA患者CK可处于正常范围,部分疾病进展较快或合并肌束颤动症状的青壮年患者则可明显升高达正常上限5倍以上[4]。电生理检查有助于将运动神经元病与肌肉疾病区分开来。研究表明,电生理检查不仅具有诊断价值,帮助定位脊髓前角或运动轴索受损[4],还能客观评估不同严重程度SMA患者的运动功能[25]。基于此,青少年成人SMA指南建议“临床诊断下运动神经元综合征,且符合下肢近端起病特点,需考虑SMA”[4]。
此外,脊柱侧凸也是SMA常见的骨科并发症。据估计,其在2型和3型SMA患者中的发病率分别为60%~90%[8]和50%[26]。SMA患者脊柱侧凸的病因主要与脊柱中轴肌肉不对称性无力萎缩相关,常伴有骨盆倾斜,逐渐引起进行性加重的脊柱侧凸。由于脊柱侧凸造成胸腔变形,进一步限制了SMA患者的呼吸能力,最终患者出现呼吸障碍加重和保持坐位困难等症状[27]。因此,国内外SMA诊疗共识建议将脊柱检查作为SMA患者常规临床评估的一部分,对于疑似或明显脊柱侧凸的患者,推荐进行前后位和侧向投照脊柱X线片检查、脊柱CT及脊柱MRI,以确定脊柱侧凸的程度及相关影响[8]。
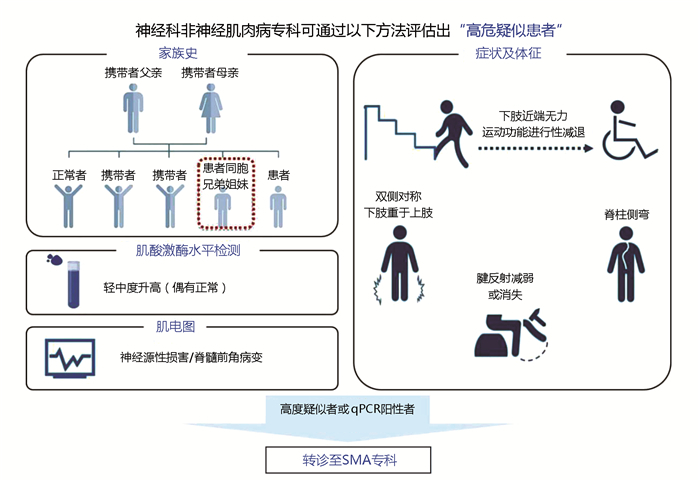
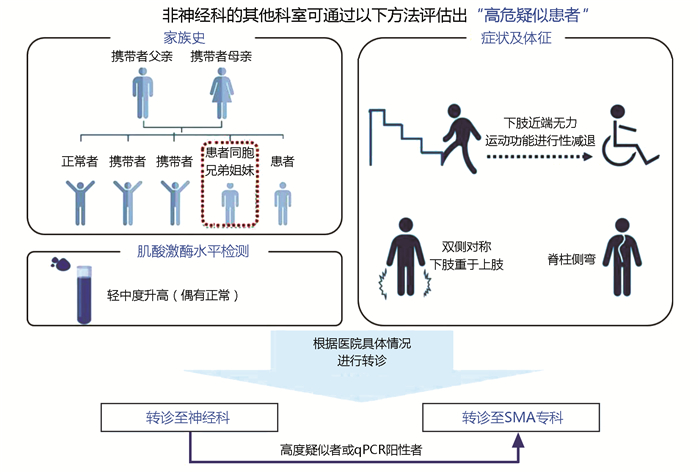
综上,基于青少年和成人SMA的临床特点,结合循证医学证据、国内外SMA诊疗指南及共识和SMA诊疗专家的讨论建议,建立起简单且清晰的青少年及成人SMA高危人群“画像”,将是一种快速高效提高诊断率、缩短诊断延迟的方法。由于在临床实践中,SMA患者很可能就诊于神经科以外的其他科室(如骨科、呼吸科、中医科、康复科、普通内科等),不同科室医生对神经肌肉病的理解程度也有所不同。因此,笔者认为这种高危“画像”的应用场景可根据就诊科室的类别与性质分层而设,即分为非神经科(图 1)和神经科非神经肌肉病专科(图 2)两类。
![]() 图 1 非神经科识别青少年及成人脊髓性肌萎缩症(SMA)高危疑似患者的“画像”qPCR:荧光定量聚合酶链式反应Figure 1. "Portrait" for identifying adolescents and adults at high-risk for spinal muscular atrophy(SMA) by non-neurologists
图 1 非神经科识别青少年及成人脊髓性肌萎缩症(SMA)高危疑似患者的“画像”qPCR:荧光定量聚合酶链式反应Figure 1. "Portrait" for identifying adolescents and adults at high-risk for spinal muscular atrophy(SMA) by non-neurologists![]() 图 2 神经科非神经肌肉病专科识别青少年及成人SMA高危疑似患者的“画像”Figure 2. "Portrait" for identifying adolescents and adults for highly suspicious of SMA by neurologists (non-neuromuscular specialists)
图 2 神经科非神经肌肉病专科识别青少年及成人SMA高危疑似患者的“画像”Figure 2. "Portrait" for identifying adolescents and adults for highly suspicious of SMA by neurologists (non-neuromuscular specialists)2.2 非神经科医生识别SMA高危疑似患者的方法
对于非神经科医生(如骨科、呼吸科、中医科、康复科等),如发现就诊患者存在以下3项中的2项临床症状:①双侧对称的下肢近端运动功能进行性减退;②下肢肌无力症状重于上肢;③脊柱侧弯。建议进一步询问其家族患病史。此时,如患者有同胞兄弟姐妹类似疾病家族史,无需做其他检查,应立即转诊至神经科(如条件允许可直接转诊至SMA专科或神经肌肉病专科)进一步检查。如无同胞病史,建议进行血CK检测,如CK水平轻中度升高(正常上限3倍以内)或正常,符合SMA特点,则转诊至神经科或神经肌肉病专科(图 1)。
2.3 神经科非神经肌肉病专科医生识别SMA高危疑似患者的方法
神经科医师接诊后,需详细询问病史,并进行神经科查体。如符合下肢起始,近端为重的对称性肢体无力萎缩,疾病逐渐进展,四肢腱反射减弱或消失,需考虑SMA的可能。如同胞中有类似患者,可不做其他辅助检查,直接行SMN1基因荧光定量聚合酶链式反应(quantitative polymerase chain reaction,qPCR)检测或其他SMN1、SMN2基因检测,如多重连接探针扩增技术(multiplex ligation-dependent probe amplifica-tion,MLPA)。如接诊医生对SMA不熟悉或医院不具备SMA基因检测能力,则可转诊至有相关诊治经验的神经肌肉病专科。如果基因检测结果不支持SMA诊断,可能是合并点突变的复合杂合突变导致SMA或其他有类似临床表现的神经肌肉病,建议患者至专科医院,完善肌电图等全面评估后选择进一步诊断策略。
如无同胞病史,则建议完善血CK检测和/或肌电图检查(具备检查条件的医院,肌电图检查应作为必须检查的项目),并根据结果判断:若CK水平轻中度升高(正常上限3倍以内)或正常,同时肌电图检查结果提示广泛神经源性损害,符合SMA所致脊髓前角及脑干运动神经核团病变特点,考虑为SMA高度疑似患者,建议直接行qPCR或MLPA检测,也可转诊至SMA专科(图 2)。
笔者认为,SMA作为罕见病,多数基层临床医生接触的机会不多;非神经肌肉病专科的医生即使通过培训掌握了疾病特点,但因日常工作中不常见,时间长了,难免遗忘。因此,简单易行的高危人群“画像”方法,更有助于持续提升SMA患者的识别率,是缩短诊断延迟的行之有效方法。未来随着临床对SMA疾病探索的深入,其高危人群“画像”也将不断完善。期待这种方法能够帮助更多非神经科或神经科非神经肌肉病专科医生提升对SMA的认识,及时给予患者基因检测帮助确诊或转诊至相关诊治医院,减少SMA患者误诊、漏诊或诊断延迟的情况,从而能够让SMA患者尽快接受规范化的治疗和管理。
3. 展望和建议
近年来,生存至青少年/成年期或在青少年/成年阶段发病的SMA患者人数不断增加,SMA已不仅是一种主要影响儿童的疾病。对于青少年和成人SMA患者,其运动功能、生活能力均会随着疾病发展而逐渐下降,严重影响患者生活质量,因此针对这一人群的早确诊、早治疗是确保最佳临床获益的关键,对患者的预后至关重要。由于该病罕见,且青少年、成人患者症状不典型,临床实践中接触患者的首诊场所常为各级医院的非神经肌肉病专科。因此,为了提高相关医生的疾病诊断能力和转诊效率,开展SMA专业知识培训、建立SMA高危人群识别系统、健全SMA转诊体系,以及完善SMA疾病管理路径等策略,均是提高青少年及成人SMA疾病诊断和管理的强有力手段。
中国近10年来的2型和3型SMA的诊断时间窗已明显缩短[16],这主要得益于对SMA认识的提高和基因检测技术在全国各级医院的不断普及。包括基因治疗在内的疾病修正治疗为青少年和成年SMA患者带来了希望,但未来仍需更多针对这些患者群体的高质量临床试验和真实世界研究,包括开展长程纵向研究以验证这些疗法的药物耐受性和长期疗效。同时,为了进一步改善患者的日常活动能力和生活质量,还需提高规范化诊疗和完善多学科管理,包括康复训练、家庭照护等。医疗服务提供者有必要加强对青少年和成年SMA患者的随访,以尽量解决其疾病进展所造成的多个器官、系统并发症对日常生活带来的不良影响。
随着人们对SMA分子遗传学及病理生理机制认识的不断加深,探索更多的生物标志物用以预测SMA表型的严重程度、指导更为精准的治疗将成为可能。另外,提升神经肌肉病相关脊柱侧凸手术普及率及开发更多新的SMA治疗靶点,也是极具潜力的研究方向,将使得SMA患者及其家庭进一步获益,拥有更好的明天。
作者贡献:赵玉英负责查询资料、组织材料和撰写文稿;朱雯华负责组织材料和修改文稿;戴毅负责确定主题、修改文稿,以及最终修订和审核定稿。利益冲突:所有作者均声明不存在利益冲突。 -
![]()
图 1 非神经科识别青少年及成人脊髓性肌萎缩症(SMA)高危疑似患者的“画像”
qPCR:荧光定量聚合酶链式反应
Figure 1. "Portrait" for identifying adolescents and adults at high-risk for spinal muscular atrophy(SMA) by non-neurologists
-
[1] Mercuri E, Sumner CJ, Muntoni F, et al. Spinal muscular atrophy[J]. Nat Rev Dis Primers, 2022, 8(1): 52. doi: 10.1038/s41572-022-00380-8
[2] Verhaart IEC, Robertson A, Leary R, et al. A multi-source approach to determine SMA incidence and research ready population[J]. J Neurol, 2017, 264(7): 1465-1473. doi: 10.1007/s00415-017-8549-1
[3] Rouault F, Christie-Brown V, Broekgaarden R, et al. Disease impact on general well-being and therapeutic expecta-tions of European type Ⅱ and type Ⅲ spinal muscular atrophy patients[J]. Neuromuscul Disord, 2017, 27(5): 428-438. doi: 10.1016/j.nmd.2017.01.018
[4] 中国研究型医院学会罕见病分会, 中国罕见病联盟, 北京罕见病诊疗与保障学会, 等. 青少年成人脊髓性肌萎缩症临床诊疗指南[J]. 罕见病研究, 2023, 2(1): 70-84. doi: 10.12376/j.issn.2097-0501.2023.01.010 [5] Grotto S, Cuisset JM, Marret S, et al. Type 0 spinal muscular atrophy: further delineation of prenatal and postnatal features in 16 patients[J]. J Neuromuscul Dis, 2016, 3(4): 487-495. doi: 10.3233/JND-160177
[6] Belter L, Jarecki J, Reyna SP, et al. The cure SMA membership surveys: highlights of key demographic and clinical characteristics of individuals with spinal muscular atrophy[J]. J Neuromuscul Dis, 2021, 8(1): 109-123. doi: 10.3233/JND-200563
[7] Ito M, Yamauchi A, Urano M, et al. Epidemiological investigation of spinal muscular atrophy in Japan[J]. Brain Dev, 2022, 44(1): 2-16. doi: 10.1016/j.braindev.2021.08.002
[8] Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: Part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care[J]. Neuromuscul Disord, 2018, 28(2): 103-115. doi: 10.1016/j.nmd.2017.11.005
[9] Interventions for spinal muscular atrophy: an impetus for newborn screening[J]. EBioMedicine, 2021, 69: 103507.
[10] Gillingwater TH. Maximising returns: combining newborn screening with gene therapy for spinal muscular atrophy[J]. J Neurol Neurosurg Psychiatry, 2021, 92(12): 1252. doi: 10.1136/jnnp-2021-327459
[11] Gregg AR, Aarabi M, Klugman S, et al. Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics(ACMG)[J]. Genet Med, 2021, 23(10): 1793-1806. doi: 10.1038/s41436-021-01203-z
[12] Zhao S, Wang W, Wang Y, et al. NGS-based spinal muscular atrophy carrier screening of 10, 585 diverse couples in China: a pan-ethnic study[J]. Eur J Hum Genet, 2021, 29(1): 194-204. doi: 10.1038/s41431-020-00714-8
[13] Salort-Campana E, Quijano-Roy S. Clinical features of spinal muscular atrophy (SMA) type 3 (Kugelberg-Welander disease)[J]. Arch Pediatr, 2020, 27(7S): 7S23-7S28.
[14] Souza PVS, Pinto WBVR, Ricarte A, et al. Clinical and radiological profile of patients with spinal muscular atrophy type 4[J]. Eur J Neurol, 2021, 28(2): 609-619. doi: 10.1111/ene.14587
[15] Lin CW, Kalb SJ, Yeh WS. Delay in diagnosis of spinal muscular atrophy: a systematic literature review[J]. Pediatr Neurol, 2015, 53(4): 293-300. doi: 10.1016/j.pediatrneurol.2015.06.002
[16] Cao Y, Cheng M, Qu Y, et al. Factors associated with delayed diagnosis of spinal muscular atrophy in China and changes in diagnostic delay[J]. Neuromuscul Disord, 2021, 31(6): 519-527. doi: 10.1016/j.nmd.2021.03.002
[17] 李林康, 陶雪怡. 我国罕见病诊疗与保障事业现状及展望[J]. 中国新药与临床杂志, 2023, 42(2): 65-70, 60. https://www.cnki.com.cn/Article/CJFDTOTAL-XYYL202302001.htm [18] Mercuri E, Deconinck N, Mazzone ES, et al. Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy(SUNFISH part 2): a phase 3, double-blind, randomised, placebo-controlled trial[J]. Lancet Neurol, 2022, 21(1): 42-52. doi: 10.1016/S1474-4422(21)00367-7
[19] Ñungo Garzón NC, Pitarch Castellano I, Sevilla T, et al. Risdiplam in non-sitter patients aged 16 years and older with 5q spinal muscular atrophy[J]. Muscle Nerve, 2023, 67(5): 407-411. doi: 10.1002/mus.27804
[20] De Vivo DC, Bertini E, Swoboda KJ, et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the Phase 2 NURTURE study[J]. Neuromuscul Disord, 2019, 29(11): 842-856. doi: 10.1016/j.nmd.2019.09.007
[21] Strauss KA, Farrar MA, Muntoni F, et al. Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: the Phase Ⅲ SPR1NT trial[J]. Nat Med, 2022, 28(7): 1390-1397. doi: 10.1038/s41591-022-01867-3
[22] Cao Y, Qu Y, Bai J, et al. Transmission characteristics of SMN from 227 spinal muscular atrophy core families in China[J]. J Hum Genet, 2020, 65(5): 469-473. doi: 10.1038/s10038-020-0730-1
[23] Cancès C, Richelme C, Barnerias C, et al. Clinical features of spinal muscular atrophy (SMA) type 2[J]. Arch Pediatr, 2020, 27(7S): 7S18-7S22.
[24] Pino MG, Rich KA, Kolb SJ. Update on biomarkers in spinal muscular atrophy[J]. Biomark Insights, 2021, 16: 11772719211035643.
[25] Kelly KM, Mizell J, Bigdeli L, et al. Differential impact on motor unit characteristics across severities of adult spinal muscular atrophy[J]. Ann Clin Transl Neurol, 2023, 10(12): 2208-2222.
[26] Catteruccia M, Vuillerot C, Vaugier I, et al. Orthopedic management of scoliosis by garches brace and spinal fusion in SMA type 2 children[J]. J Neuromuscul Dis, 2015, 2(4): 453-462.
[27] Garg S. Management of scoliosis in patients with Duchenne muscular dystrophy and spinal muscular atrophy: a literature review[J]. J Pediatr Rehabil Med, 2016, 9(1): 23-29.
 下载:
下载:
计量
- 文章访问数: 113
- HTML全文浏览量: 17
- PDF下载量: 27


